Fabry

Discover Fabry Disease:
The Role of the Cardiologist
Fabry Disease Can Result in Substantial Heart Damage1
Fabry disease is a rare, progressive, and potentially life-threatening disorder that starts in early childhood and affects men and women.1-3
As an X-linked lysosomal storage disorder that is multisystemic, Fabry disease is caused by complete or partial deficiency of the lysosomal enzyme α-GAL A, leading to GL-3 and lyso-GL-3 accumulation that can result in damage to the heart, kidneys, and cerebrovascular system.2,3
Early Diagnosis and Management Are Critical as Fabry Disease Can Decrease Life Expectancy Due to Heart and Organ Damage2,4-6
.webp)
.webp/jcr:content/img-4%20(4).webp)
Understanding Fabry Disease
Worldwide Incidence7
1 in 40,000
FD is an X-linked disease caused due to deficiency of α-galactosidase A. An average diagnostic delay of 15–18 years is seen in FD.
Test for diagnosis
- Enzyme assays to detect α-Gal activity
- Genetic testing to confirm diagnosis
Cardiac Disease is the Leading cause of Death in Patients with Fabry Disease1
Consider Fabry Disease in your Differential Diagnosis when10:
Presentation: Unexplained HCM and/or LVH, fibrosis in the lateral wall of the left ventricle, low native T1 values, elevated hs-troponin in the absence of acute event9,11
Family history: Cardiomyopathy, premature stroke, sudden cardiac death, and renal failure10,12
ECG: Bradycardia, short PR interval (young age), AV block (cardiac clinical manifestations in the 3rd decade of life onward), voltage criteria for LVH, ST segment changes, T-wave inversion8,10
If you see Unexplained Cardiomyopathy, Consider Fabry Disease.13
Cardiologists have the tools to screen for, diagnose, and manage Fabry disease.
| Testing is Straightforward8,14,15 | In patients with unexplained HCM, conduct genetic testing that includes the GLA gene. | Treatment is Available for your Patients with Fabry Disease | |
| Males | α-GAL A enzyme assay and/or GLA gene sequencing | ||
| Females | GLA gene sequencing Affected females may have normal to low enzyme activity in plasma or leukocytes; therefore, GLA gene sequencing is required to confirm diagnosis in females. | Stabilizing disease progression is an important goal.8 | |
Damage to cardiac, renal, and cerebrovascular systems can have life-threatening impact. Monitor patients in accordance with published management guidelines and clinicians’ medical expertise.8
α-GAL A, α-galactosidase A; AV, atrioventricular; CNS, central nervous system; ECG, electrocardiogram; GLA, galactosidase-alfa; HCM, hypertrophic cardiomyopathy; hs, high-sensitivity; LVH, left ventricular hypertrophy.
- Azevedo O, et al. Int J Mol Sci. 2021;22(9):4434.
- Lidove O, et al. Clin Genet. 2012;81(6):571-577.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49.
- MacDermot KD, et al. J Med Genet. 2001;38(11):750-760.
- MacDermot KD, et al. J Med Genet. 2001;38(11):769-775.
- Waldek S, et al. Genet Med. 2009;11(11):790-796.
- Cainelli F, et al. Front Genet. 2021;12:657824.
- Ortiz A, et al. Mol Genet Metab. 2018;123(4):416-427.
- Thompson RB, et al. Circ Cardiovasc Imaging. 2013;6:637-645.
- Yousef Z, et al. Eur Heart J. 2013;34(11):802-808.
- Seydelmann N, et al. J Am Heart Assoc. 2016;5:e002839.
- Baig S, et al. Europace. 2018;20(FI2):f153-f161.
- Hagège A, et al. Arch Cardiovasc Dis. 2019;112(4):278-287.
- Biegstraaten M, et al. Orphanet J Rare Dis. 2015;10:36.
- Bales ND, et al. Pediatr Cardiol. 2016;37(5):845-851.
Discover Fabry Disease:
The Role of the Nephrologist
Renal Manifestations of Fabry Disease are common and can be Life Threatening.1,2
Fabry disease is a rare, progressive, and potentially life-threatening disorder that starts in early childhood and affects men and women.2-4
As an X-linked lysosomal storage disorder that is multisystemic, Fabry disease is caused by complete or partial deficiency of the lysosomal enzyme α-GAL A, leading to GL-3* and lyso-GL-3 accumulation that can result in damage to the kidneys, heart, and cerebrovascular system.4
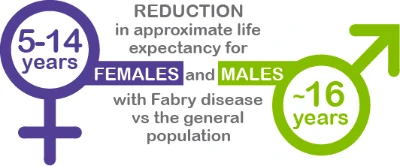
Early Diagnosis and Management are Critical as Fabry Disease can Decrease Life Expectancy due to Organ Damage.4-7
.png)
.png/jcr:content/img-4%20(1).png)
Understanding Fabry Disease
Worldwide Incidence7
1 in 40,000
FD is an X-linked disease caused due to deficiency of α-galactosidase A. An average diagnostic delay of 15–18 years is seen in FD.
Test for diagnosis
- Enzyme assays to detect α-Gal activity
- Genetic testing to confirm diagnosis
Renal Manifestations are a Prominent Indication of Fabry Disease and can be Life Threatening10
Presentation: Microalbuminuria, proteinuria, reduced GFR, hematuria (rare), renal cysts (mainly parapelvic), and (in early disease) reduced systemic BP and glomerular hyperfiltration11
Family history: Cardiomyopathy, premature stroke, sudden cardiac death, and renal failure12,13
Laboratory and Biopsy Findings
Urinalysis: “Maltese cross,” “urinary mulberry cells,” biomarkers GL3* and lyso-GL311
Biopsy: Glomerulosclerosis, vascular lesions, vacuolation, and lysosomal GL3 deposits11
Rule out Fabry disease in your patients with unexplained CKD.
| Renal Best Practice Screening Recommendations14 | Testing is Straightforward9,15 | |
| Males | Screening MALES <50 years of age with unexplained CKD | α-GAL A enzyme assay and/or GLA gene sequencing |
| Females | Screening FEMALES at any age with unexplained CKD and other symptoms associated with Fabry disease | GLA gene sequencing Affected females may have normal-to-low enzyme activity in plasma or leukocytes; therefore, GLA gene sequencing is required to confirm diagnosis in females. |
Damage to renal, cardiac, and cerebrovascular systems can have a life-threatening impact. Monitor patients in accordance with published management guidelines and clinicians’ medical expertise.9
*GL-3 and Gb3 are interchangeable terms for globotriaosylceramide.
BP, blood pressure; CKD, chronic kidney disease; GFR, glomerular filtration rate; GLA, galactosidase-alfa.
- Waldek S, et al. BMC Nephrol. 2014;15:72.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49.
- Azevedo O, et al. Int J Mol Sci. 2021;22(9):4434.
- Lidove O, et al. Clin Genet. 2012;81(6):571-577.
- MacDermot KD, et al. J Med Genet. 2001;38(11):750-760.
- MacDermot KD, et al. J Med Genet. 2001;38(11):769-775.
- Waldek S, et al. Genet Med. 2009;11(11):790-796.
- Cainelli F, et al. Front Genet. 2021;12:657824.
- Ortiz A, et al. Mol Genet Metab. 2018;123(4):416-427.
- Schiffmann R, et al. Kidney Int. 2017;91(2):284-293.
- Silva CAB, et al. Can J Kid Health Dis. 2021;8:1-14.
- Baig S, et al. Europace. 2018;20(FI2):f153-f161.
- Yousef Z, et al. Eur Heart J. 2013;34(11):802-808.
- Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505-517.
- Biegstraaten M, et al. Orphanet J Rare Dis. 2015;10:36.
