- Article
- Source: Campus Sanofi
What is Fabry disease?
.webp/jcr:content/herobanner_what_is_fabry%20(1).webp)
14 Male Years | 19 Female Years
The average time to diagnosis in Fabry disease is 14 years for males and 19 years for females4
Fabry disease is an X-linked lysosomal storage disorder that affects men, women, and children of all ethnicities.1,2 Estimates of the prevalence of Fabry disease range from a newborn screening study which found approximately 1 in 4,600 males were affected (classical and non classical) to the more typically reported prevalence of 1 in 40,000 males with an unknown number in females.3
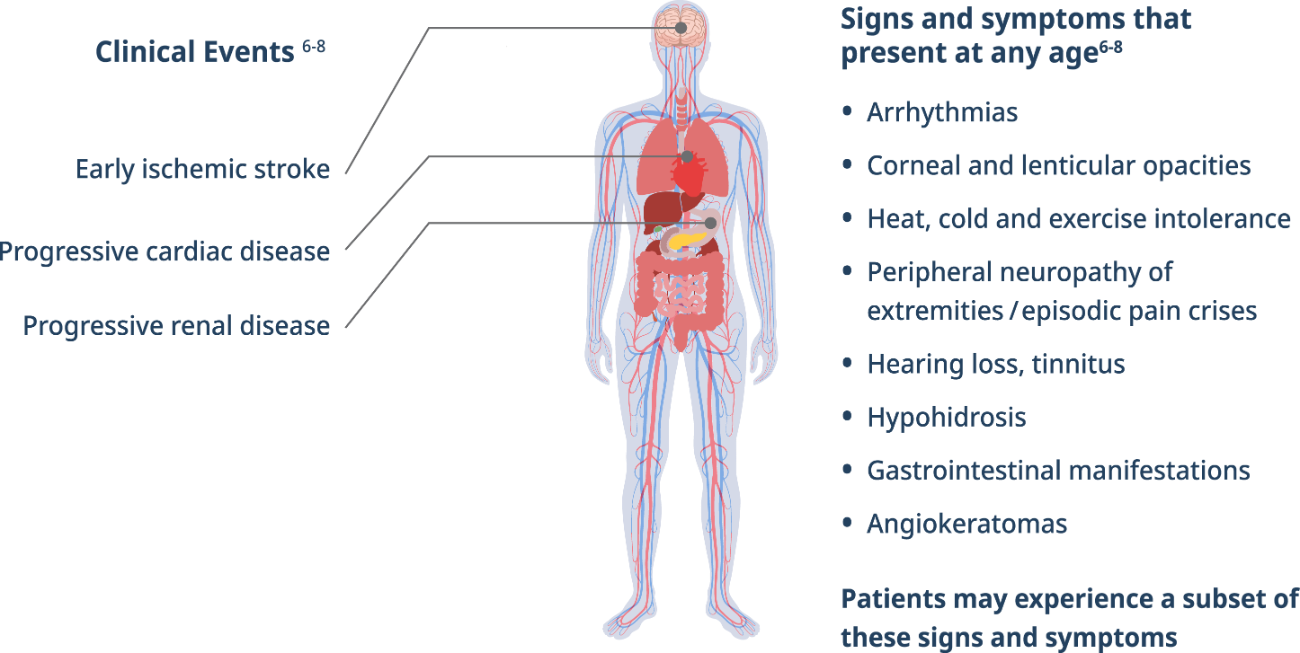
Fabry is a progressive, multisystemic disorder that can result in potentially life-threatening damage to the kidney, heart, and brain.4
Early signs and symptoms of Fabry disease, which can start as early as childhood or adolescence, may include pain, gastrointestinal disturbances, angiokeratomas, hypohidrosis, proteinuria, and other signs and symptoms.5As Fabry disease progresses, renal function deterioration and hypertrophic cardiomyopathy may occur, and the risk of developing fatal complications such as end-stage renal disease, stroke, cardiac fibrosis, arrhythmias, and premature death increases.5
Signs and symptoms diagram
.2023-10-24-08-09-50.png)
Road to diagnosis
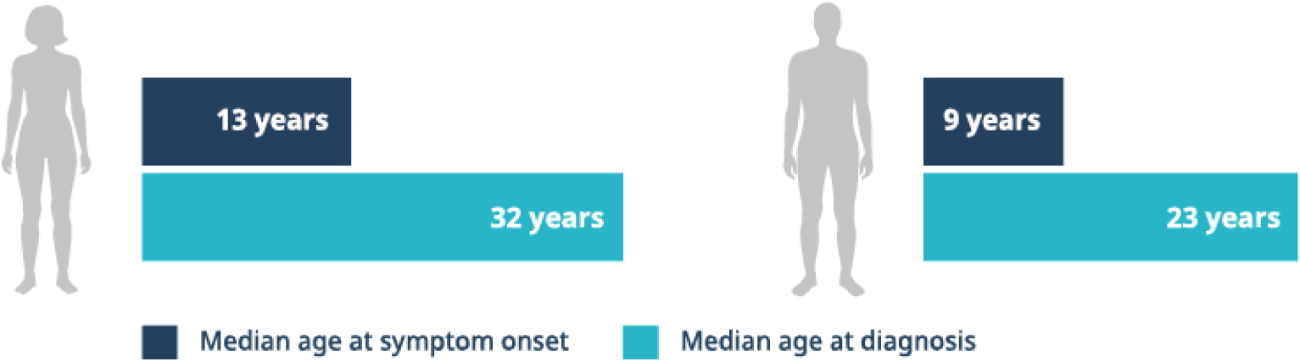
In a study based upon the Fabry Registry, due to the nonspecific and heterogeneous nature of early symptoms in Fabry disease, diagnostic delays and misdiagnosis were common.4
Fabry disease is often misdiagnosed and confused with rheumatoid or juvenile arthritis, rheumatic fever, erythromelalgia, Raynaud’s syndrome, neurosis, lupus, acute appendicitis, or multiple sclerosis. Pain, particularly in children, can be dismissed as malingering.5
Connecting seemingly unrelated symptoms to Fabry disease can help avoid diagnostic delays and help patients receive disease management sooner.4
Importance of Screening for Fabry Disease in High-Risk Populations
Although Fabry is considered a rare disease, the prevalence of Fabry disease in patients with certain disorders, such as unexplained chronic kidney disease and hypertrophic cardiomyopathy, may be higher than in the general population.10,11 Consequently, it is important to screen patients with these conditions for Fabry disease.
Delays in diagnosis can be substantial4
.2023-10-24-08-10-45.png)
Pathophysiology
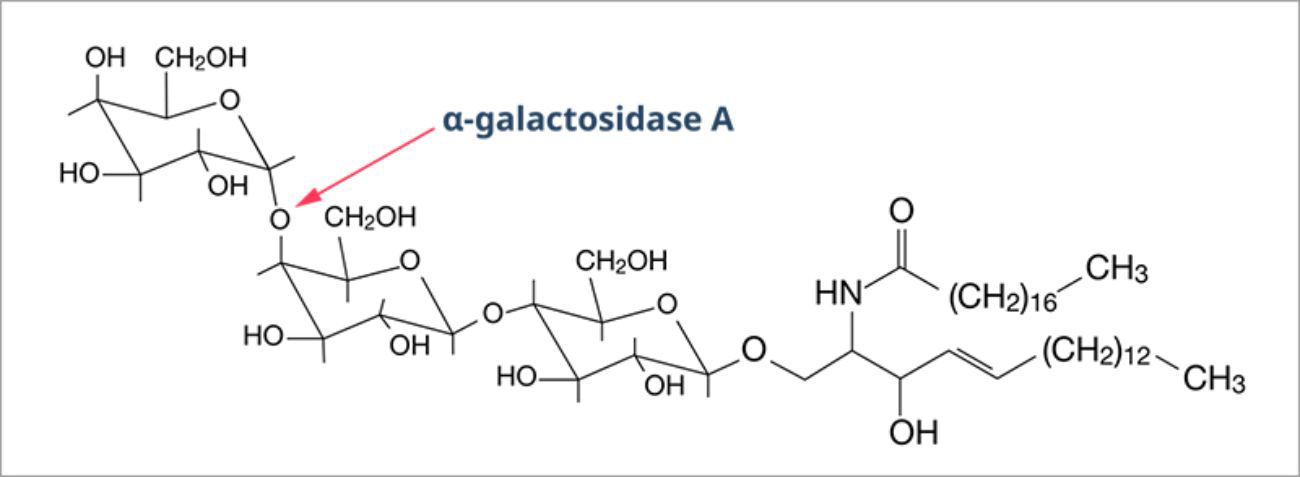
In Fabry disease, mutations in the GLA gene, located on the X chromosome, result in defects in the synthesis and/or function of alpha-galactosidase A (α-Gal A).12 α-Gal A is a lysosomal enzyme, which usually metabolizes globotriaosylceramide (also known as GL-3 or Gb3) and prevents it from accumulating.9
Complete or partial deficiency of α-Gal A leads to progressive accumulation of glycosphingolipids, particularly GL-3, in the lysosomes of numerous cell types.5
In Fabry disease, accumulation of GL-3 begins prenatally, or in utero, and continues over a lifetime.1,2 With advancing age, the progressive lysosomal GL-3 accumulation – particularly in the microvasculature – leads to renal failure, heart disease, stroke, and premature demise, typically in the fourth or fifth decade of life.4,5,9,13
.2023-10-24-08-11-33.png)
The broad spectrum of symptoms in Fabry disease are as a result of a patient’s residual α-Gal A
- Desnick RJ, et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2014:1–64.
- Desnick RJ. Ann Intern Med. 2003;138(4):338–346.
- Laney DA, et al. J Genet Couns. 2008;17(1):79–83.
- Eng CM, et al. J Inherit Metab Dis. 2007;30(2):184–192.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1–49
- Barba-Romero MA, et al. Int J Clin Pract. 2011;65(8):903–910.
- Mehta A. QJM. 2002;95(10):647–653.
- Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112–128.
- Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600–1607.
- Terryn W et al. Nephrol Dial Transplant. 2013; 28:505–517.
- Elliot PM, et al. Eur Heart J. 2014; 35 (39): 2733–2779.
- Nagueh SF. Circulation. 2014;130(13):1081–1090
- Sims K. et al. Stroke. 2009:40(3):788–794.
