- Article
- Source: Campus Sanofi
What is Pompe disease?
Pompe disease (glycogen storage type II disease) is caused by a deficiency of acid alphaglucosidase (GAA) enzyme activity, resulting in lysosomal glycogen accumulation in muscles and irreversible muscle damage.1-3
About Pompe disease
Pompe disease most commonly affects the respiratory and musculoskeletal muscles.1
Patients with Pompe disease will usually present with a broad spectrum of clinical phenotypes at different ages:2
- As infants, also known as infantile-onset Pompe disease (IOPD)
- As children and adults, which is also known as late-onset Pompe disease (LOPD)
Pompe disease is also known as: acid maltase deficiency or glycogen storage type II disease (GSD II).1
Pompe disease is a life-limiting, progressive neuromuscular disorder caused by an inherited deficiency of enzyme activity leading to irreversible muscle damage1,2,5,6 – but enzyme replacement therapy is available7
Illustration based on Thurberg, et al. 2006.4

Epidemiology
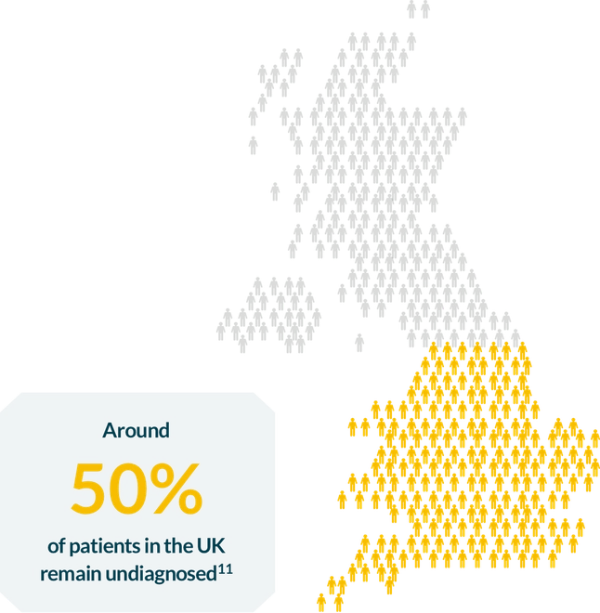
Incidence estimates for Pompe disease range from 1 in 33,333 to 1 in 138,0008-9. However, it is difficult to know exactly how many people are actually affected.
It is estimated that the current worldwide prevalence may be 1 in 57000.10
Genetics and inheritance
A gene located on chromosome 17 encodes for the production of acid alpha-glucosidase (GAA), the enzyme responsible for breaking down glycogen to glucose inside lysosomes.12 A genetic mutation within this gene causes a deficiency, or complete absence, of GAA enzyme which results in intra-lysosomal accumulation of glycogen, primarily in muscle cells.13 Pompe disease is an inherited autosomal recessive disease: two faulty genes must be inherited from both parents for the person to develop Pompe disease.14
At 1 in 57,000 people10 there are estimated to be other 1,000 people with Pompe in the UK alone.
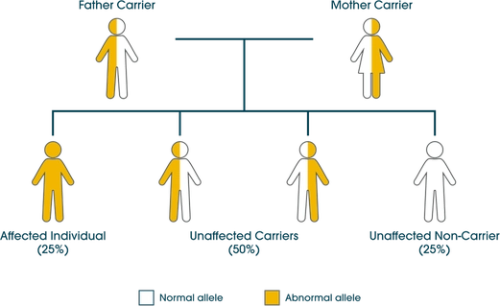
Illustration based on Thurberg, et al. 2006.4
- Hirschhorn R, et al. The Metabolic and Molecular Basis of Inherited Disease 2001;8(3):3389–3420
- American Association of Neuromuscular & Electrodiagnostic Medicine. Muscle Nerve 2009; 40(1):149–160
- Fukada T, et al. Mol Ther. 2006;14(6):831–839
- Thurberg BL, et al. Lab Invest. 2006;86(12):1208–1220
- Hagemans ML, et al. Neurology 2005;64(12):2139–2141
- Kishnani PS, et al. Genet Med. 2006; 8(5):267–288
- Myozyme Summary of Product Characteristics. Accessed August 2022
- Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan Screening Program. Pediatrics 2008; 122: e39–45.
- Ausems MGEM, Verbiest J, Hermans MMP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in the Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999; 7: 713–716.
- AGSD, Pompe Disease (GSD2). Website, available online here: https://agsd.org.uk/all-about-gsd/gsd-variants/pompe-disease-gsd2 / [Last accessed October 2019]
- Ausems MGEM, Lochman P, van Diggelen OP, Ploos van Amstel HK, Reuser AJJ, Wokke JHJ. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 1999; 52: 851–853.
- Reuser AJ, Hirschhorn R, Kroos MA. Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. eds. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill; 2019. Accessed August, 2022. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225890450
- Pittis MG, et al. Acta Myol. 2007;26(1):67–71
- Pompe Disease - NORD (National Organization for Rare Disorders). Website, available here: https://rarediseases.org/rare-diseases/pompe-disease/ [Last accessed October 2019]
.webp/jcr:content/signs-and-symptoms%20(1).webp)
Signs and Symptoms of Pompe disease
Pompe disease should be considered as part of the differential diagnosis for all children and adults presenting with limb-girdle muscle weakness and respiratory insufficiency1