
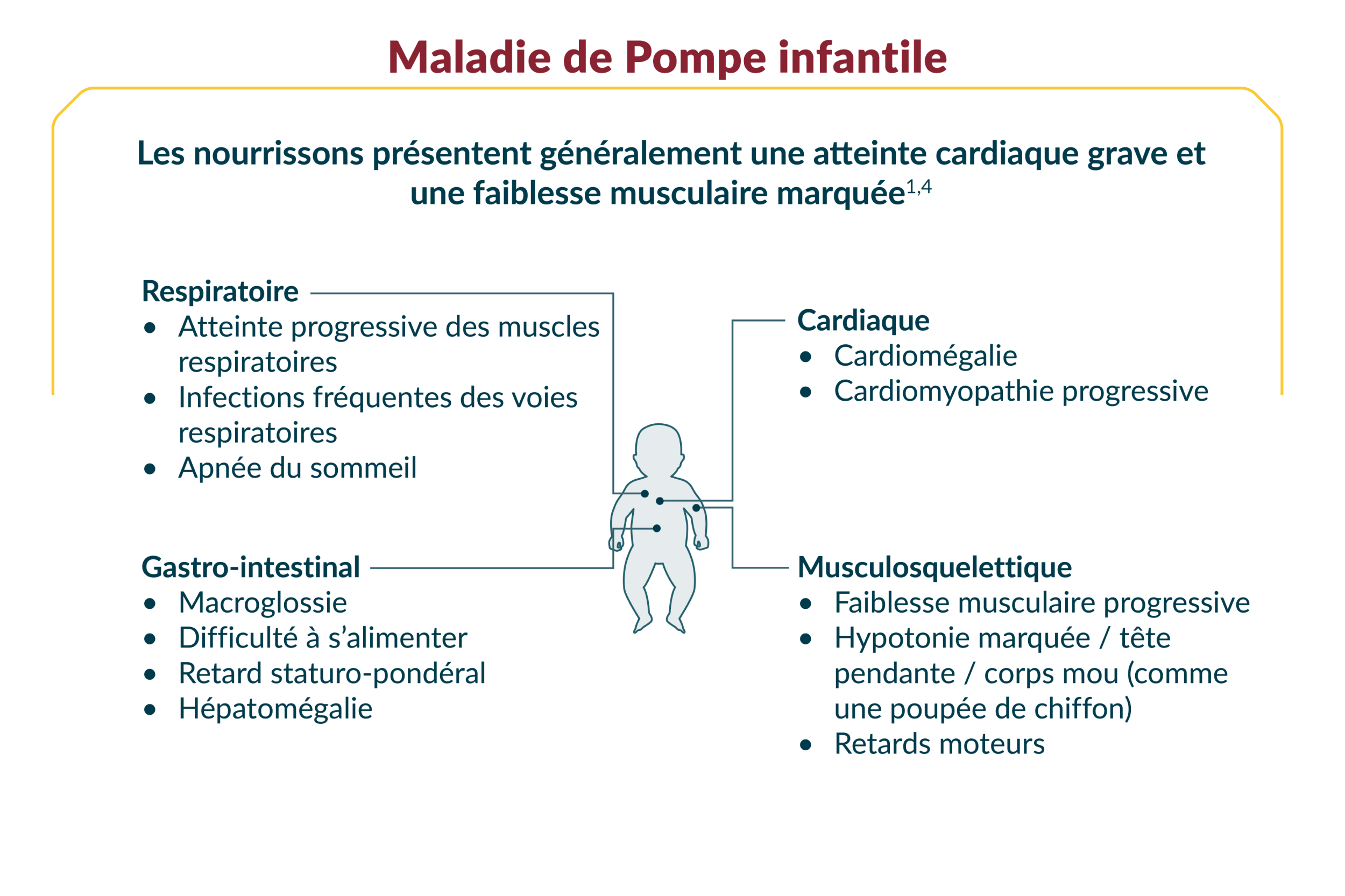
La maladie de Pompe
La maladie de Pompe (MP) est un trouble neuromusculaire évolutif rare potentiellement mortel qui atteint plusieurs organes et systèmes et qui touche principalement les muscles cardiaque, squelettiques et lisses1,2
- La maladie de Pompe est un trouble génétique à transmission autosomique récessive résultant d’un déficit en α-glucosidase acide (GAA), une enzyme lysosomale, ou d'un dysfonctionnement de celle-ci1.
- Un déficit en GAA entraîne l'accumulation de glycogène dans les lysosomes de nombreux tissus, ceux du muscle cardiaque et des muscles squelettiques étant les plus gravement atteints1.
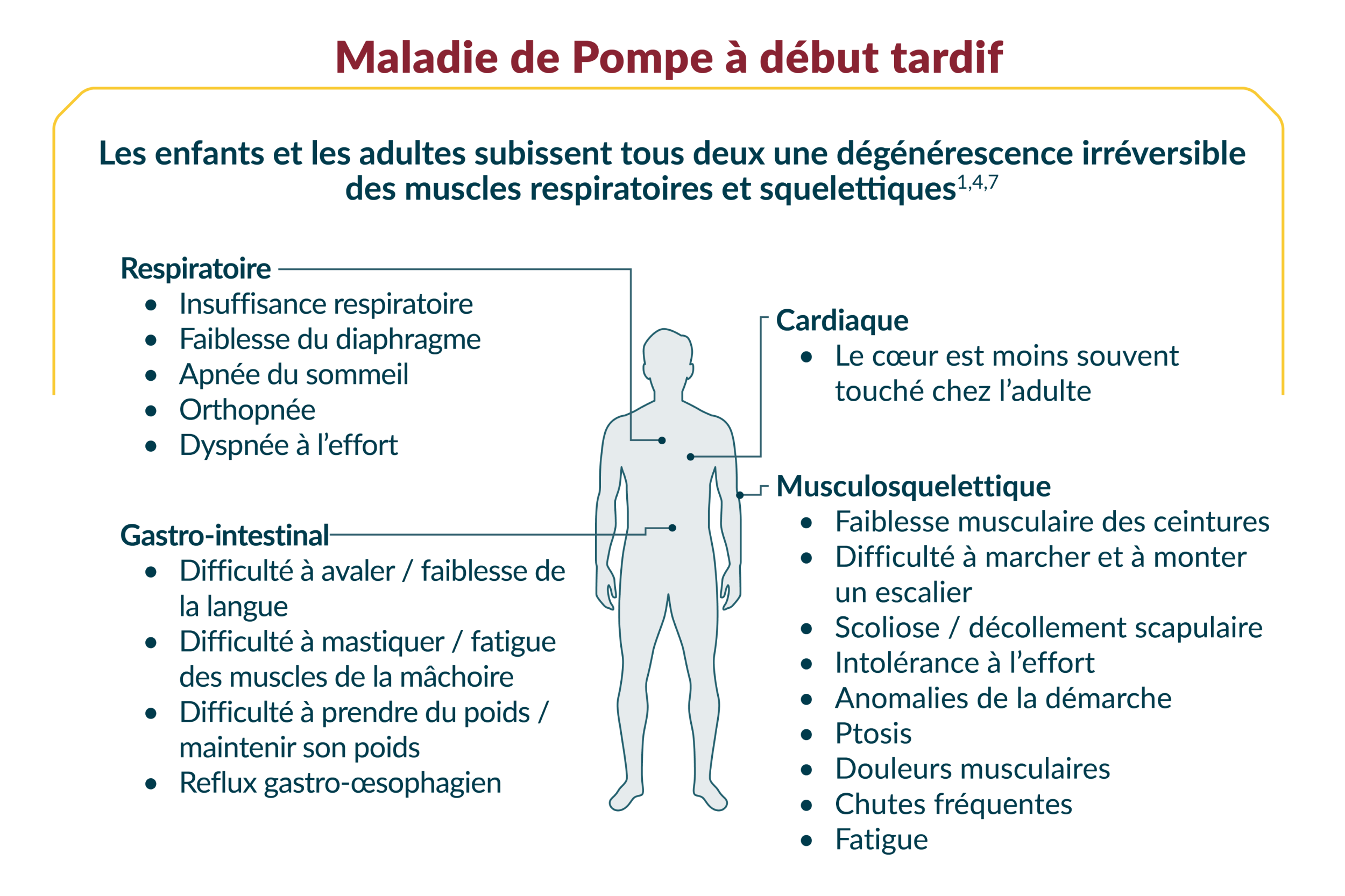
- La maladie de Pompe se présente sous deux grands phénotypes : la maladie de Pompe infantile et la maladie de Pompe à début tardif1.
Les patients présentent habituellement des symptômes touchant quatre systèmes d’organes : musculosquelettique, respiratoire, cardiaque et gastro-intestinal1


Quels sont les symptômes?
La maladie de Pompe touche de nombreux organes et peut être difficile à diagnostiquer1,5. Soyez attentifs à l’apparition des symptômes et des manifestations de la maladie de Pompe.
Les nourrissons atteints de MP infantile et les enfants et les adultes atteints de MP à début tardif ne présentent pas les mêmes symptômes6
Lors d’une étude menée auprès de 44 patients (27 femmes et 17 hommes) ayant reçu un diagnostic de maladie de Pompe à début tardif à l’Institut Friedrich-Baur entre 1985 et 20118 :
77 %
des patients atteints de la maladie de Pompe à début tardif présentent une intolérance à l’effort et une faiblesse des muscles des ceintures
55 %
des patients atteints de la maladie de Pompe à début tardif présentent une insuffisance respiratoire (pouvant avoir des répercussions sur le sommeil) et une faiblesse des muscles des ceintures
D’après Schüller A, et al.
La maladie de Pompe à début tardif entraîne, bon an mal an, un risque plus important de fardeau majeur pour les patients qui en sont atteints9,10
D’après un projet de recherche en cours mené par l’International Pompe Association auprès de 255 patients de plus de 2 ans atteints de la maladie de Pompe à début tardif10 :
+13 %
Le risque d’avoir besoin d’un fauteuil roulant augmente de 13 % chaque année qui suit le diagnostic (p < 0,001)
+8 %
Le risque d’avoir besoin d’assistance respiratoire augmente de 8 % chaque année qui suit le diagnostic (p < 0,001)
D’après Hagemans ML, et al.
Si vous soupçonnez la présence de la maladie de Pompe chez l’un de vos patients, agissez.
En savoir plus sur les tests diagnostiques de la maladie de Pompe
Quel est le mode de transmission de la maladie de Pompe?
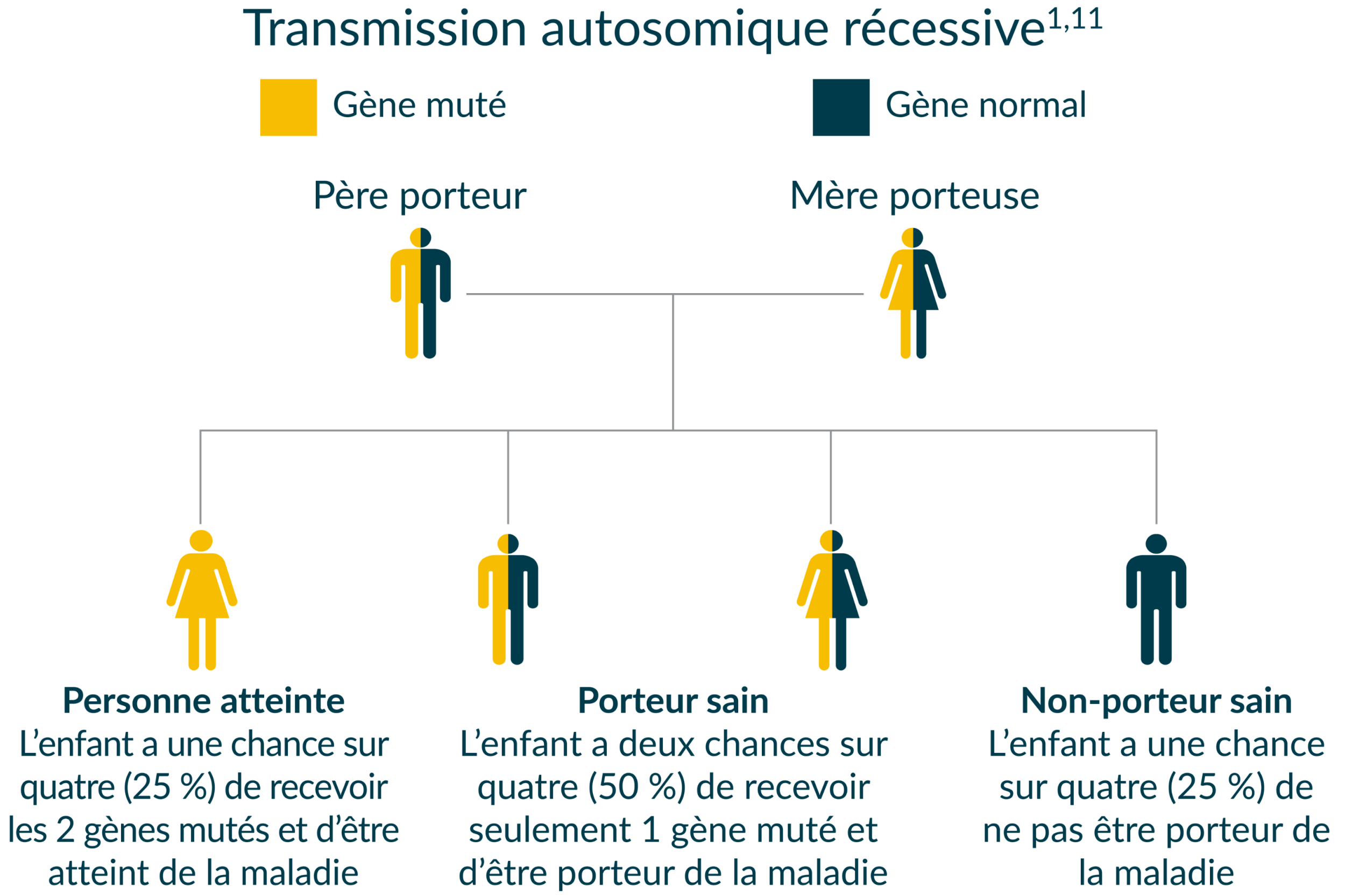
L’un de vos patients a reçu un diagnostic de maladie de Pompe? Dépistez la maladie chez les membres de sa famille
Il est important de poser un diagnostic précoce. L’analyse génétique de la mutation chez les membres de la famille permet de repérer d’autres patients et porteurs de la maladie de Pompe1.
Sanofi offre gratuitement le dosage de l’activité de l’enzyme GAA et le séquençage génétique aux patients chez qui la maladie de Pompe est soupçonnée.
En savoir plus sur les tests diagnostiques de la maladie de Pompe
Quelles sont certaines des considérations cliniques?
Cherchez-vous à dépister la maladie de Pompe?
Vous jouez un rôle important pour repérer les patients atteints de cette maladie évolutive qui est souvent mortelle.
![]()
Un diagnostic de maladie de Pompe peut être envisagé
chez les patients qui présentent les phénotypes suivants12 :
1er phénotype : patients présentant une faiblesse musculaire proximale ou axiale, avec ou sans symptômes respiratoires
2e phénotype : patients atteints d’une insuffisance respiratoire restrictive
3e phénotype : patients atteints d’une hyperCKémie asymptomatique*
- Lors d’une étude menée auprès de 3 076 adultes atteints d’hyperCKémie et / ou de faiblesse musculaire des ceintures, le séquençage génétique a permis d’établir la prévalence de la maladie de Pompe à 2,4 %13
- Lors d’une étude menée auprès de 275 patients atteints d’hyperCKémie et / ou de dystrophie musculaire des ceintures, le séquençage génétique a permis d’établir la prévalence de la maladie de Pompe à 3,6 %14
Envisageriez-vous de dépister la maladie de Pompe face à ces signes et symptômes courants?
Pour en savoir plus, téléchargez cette liste de vérification à utiliser lors d’un diagnostic différentiel
Examens et symptômes additionnels1,12,15 :
Tracé myopathique à l’EMG
- Signes d’activité spontanée et de décharges myotoniques
- Souvent constatés dans les muscles paravertébraux
Examen physique général
- Anomalies squelettiques telles qu’une scoliose, une hyperlordose lombaire et une rigidité de la colonne vertébrale
Symptômes liés à une atteinte cardiaque
- Arythmies cardiaques, hypertrophie ventriculaire et syndrome de Wolf-Parkinson-White
Symptômes liés à une insuffisance respiratoire
- Dyspnée et apnée obstructive du sommeil
- Antécédents d’infections respiratoires
- Céphalées matinales
- Somnolence diurne excessive
- Toux faible ou diminution de l’efficacité de la toux
Les atteintes aux autres organes peuvent comprendre une perte d’audition neurosensorielle, des anomalies vasculaires parmi lesquelles des anévrismes cérébraux, une atteinte gastro-intestinale avec macroglossie, hépatomégalie, diarrhée et un faible indice de masse corporelle.
CK : créatine kinase.
* Entre 1,5 et 15 fois les taux normaux; ~300 à 2 000 U/L.
Comment la maladie de Pompe est-elle diagnostiquée?
Soyez à l'affût de la maladie de Pompe
Le diagnostic à un stade précoce de la maladie permet de mettre en place plus rapidement une intervention thérapeutique et une prise en charge du patient.
- Les symptômes de la maladie de Pompe peuvent s’apparenter à ceux d'autres maladies, ce qui retarde le diagnostic1,5,16
- Le délai médian avant un diagnostic définitif de la maladie de Pompe à début tardif peut aller jusqu’à 7 ans5
Diagnostics différentiels pour les maladies neuromusculaires

† La liste des signes et symptômes énumérés dans ce tableau n’est pas exhaustive et ne comprend pas tous les signes et symptômes qui pourraient survenir chez une personne atteinte de ces maladies.
Moyens de dépistage de la maladie de Pompe
Dosage de l’activité enzymatique35
Il s'agit d'un test non invasif relativement rapide qui est utilisé lorsque l’on souhaite écarter ou confirmer le diagnostic de maladie de Pompe spécifiquement. Le patient fournit une goutte de sang séché, qui est analysée afin de mesurer le niveau d’activité de l’enzyme alpha-glucosidase acide (GAA). Si l’activité enzymatique est faible, le même échantillon de sang peut être utilisé pour le séquençage monogénique du gène GAA afin de confirmer le diagnostic de maladie de Pompe.
Séquençage génétique36
Le séquençage génétique, effectué par séquençage à haut débit, appuie le diagnostic différentiel en permettant d’évaluer rapidement et simultanément diverses mutations génétiques associées à des troubles dont les symptômes recoupent ceux de la maladie de Pompe. Une analyse génétique visant à dépister les troubles musculaires, comme celle offerte par Sanofi, permet d'évaluer plus de 100 mutations génétiques distinctes à partir d'un seul échantillon prélevé par frottis buccal. Ce type de séquençage permet de mettre au jour des mutations connues et peut aussi être utilisé pour confirmer les résultats d’un dosage de l’activité enzymatique.
Sanofi offre gratuitement le dosage de l’activité de l’enzyme GAA et le séquençage génétique aux patients chez qui la maladie de Pompe est soupçonnée.
Pour obtenir plus d’information, envoyez-nous un courriel à cette adresse.
Considérations pour la prise en charge continue de la maladie de Pompe1,15,37,38

Chez les patients atteints de la maladie de Pompe, des soins multidisciplinaires précoces et continus sont des éléments clés de la prise en charge des symptômes et d’une meilleure issue1,2,15
Centres d’expertise neuromusculaire et sur la maladie de Pompe
Vous pouvez aussi orienter votre patient vers un centre d’expertise régional pour le diagnostic, le traitement et le suivi de la maladie de Pompe.
Cliquez sur votre région pour obtenir les coordonnées des centres de votre province.
Centres d’expertise neuromusculaire et sur la maladie de Pompe en Alberta
CALGARY
Dr Aneal Khan
Personne-ressource : clinic@magiccalgary.ca
Téléphone : 587 885-3158
Télécopieur : 587 441-8380
Clinique MAGIC (Metabolics and Genetics in Calgary)
215–971 64th Ave. NE
Calgary (Alberta)
T2E 7Z4
Dr Sameer Chhibber
Téléphone : 587 747-5615
Télécopieur : 587 747-5616
Alberta Neurologic Clinic
Suite 300, 1608 17th Ave. SW
Calgary (Alberta)
T2T 0E3
EDMONTON
Dre Shailly Jain
Personne-ressource : shailly.jain@albertahealthservices.ca
Téléphone : 780 407-7333
Télécopieur : 780 407-6845
Clinical and Metabolic Genetics Clinic
Service de génétique médicale (Department of Medical Genetics)
Université de l’Alberta
8–53 Medical Sciences Building
8613 114 Street
Edmonton (Alberta)
T6G 2H7
Dre Cecile Phan
Personne-ressource : phan@ualberta.ca
Téléphone : 780 248-1698
Télécopieur : 780 407-1507
Kaye Edmonton Neuromuscular Clinic
4C–101, 11400 University Ave. NW
Edmonton (Alberta)
T6G 2G3
Centres d’expertise neuromusculaire et sur la maladie de Pompe en Colombie-Britannique
VANCOUVER
Dre Michelle Mezei
Personne-ressource : mezei@mail.ubc.ca
Adult Metabolic Diseases Clinic
2775 Laurel Street
Vancouver (Colombie-Britannique)
Télécopieur : 604 875-5967
Dre Anna Lehman
Personne-ressource : anna.lehman@vch.ca
Téléphone : 604 875-5965
Adult Metabolic Diseases Clinic
2775 Laurel Street
Vancouver (Colombie-Britannique)
Télécopieur : 604 875-5967
Centres d’expertise neuromusculaire et sur la maladie de Pompe en Ontario
HAMILTON
Dr Mark Tarnopolsky
Téléphone : 905 521-7933
Télécopieur : 905 521-2638
Département de médecine
Clinique sur les maladies neuromusculaires
McMaster University H S C 2H22
1200 Main Street
Hamilton (Ontario)
L8N 3Z5
LONDON
Dre Anita Florendo-Cumbermack
Téléphone : 519 663-3129
Secrétaire : Sue Robinson
Télécopieur : 519 663-3328
London Health Sciences Centre
339 Windermere Road
London (Ontario)
N6A 5A5
OTTAWA
Dr Pierre Bourque
Téléphone : 613 761-4777
Assistante administrative : Janine Brooks-Jean
Courriel : jabrooks@toh.ca
Télécopieur : 613 761-5403
Hôpital d’Ottawa
1053, avenue Carling
Ottawa (Ontario)
K1Y 4E9
Centres d’expertise neuromusculaire et sur la maladie de Pompe au Québec
MONTRÉAL
Médecins expérimentés dans le traitement de la maladie de Pompe
Dre Genge – Neurologue
Dre O’Ferrall – Neurologue
Dr Blanchard – Neurologue
Dr Massie – Neurologue
Personne-ressource :
Josée Terrigno
514 398-8551
Institut-Hôpital neurologique de Montréal (Le Neuro)
QUÉBEC
Médecins expérimentés dans le traitement de la maladie de Pompe
Dr Brunet – Neurologue
Personne-ressource :
Manon Gravel – Infirmière clinicienne
clinique-neuromusculaire@chuquebec.ca
418 525-4444 poste 66893
Hôpital de l’Enfant-Jésus
SHERBROOKE
Médecins expérimentés dans le traitement de la maladie de Pompe
Dr Levesque – Génétique
Dre Lareau-Trudel – Neurologue
Personne-ressource :
Caroline Barr – Infirmière clinicienne en génétique
caroline.barr.ciussse-chus@ssss.gouv.qc.ca
819 346-1110, poste 16828
Hôpital Fleurimont
- Kishnani PS, et al. Genet Med 2006;8(5):267–88.
- American Association of Neuromuscular & Electrodiagnostic Medicine. Muscle Nerve 2009;40(1):149–160.
- Kishnani PS, et al. J Pediatr 2006;148(5):671–76.
- Kishnani PS, et al. J Pediatr 2004;144:S35–43.
- Winkel LPF, et al. J Neurol 2005;252(8):875–84.
- Hirschhorn R, et al. Dans : Scriver CR, et al., rédacteurs. The Metabolic & Molecular Bases of Inherited Disease. 8e éd. New York, NY: McGraw-Hill; 2001:3389–420.
- Chan J, et al. Molecular Genetics and Metabolism 2017;120(3):163–72.
- Schüller A, et al. Dans : Am J Med Genet (Part C: Seminars in Medical Genetics) 2012;160(1):80–8. Hoboken: Wiley Subscription Services, Inc.; A Wiley Company.
- Rigter T, et al. Molecular Genetics and Metabolism 2012;107(3):448–55.
- Hagemans ML, et al. Neurology 2005;64(12):2139–41.
- Leslie N, Bailey L. Dans : Adam MP, et al., rédacteurs. GeneReviews 2007 [Mise à jour du 2 novembre 2023]. Seattle, WA: University of Washington.
- Toscano A, et al. Acta Myologica 2013;32(2):78.
- Lukacs Z, et al. Neurology 2016;87(3):295–98.
- Savarese M, et al. Neuromuscul Disord 2018;28(7):586–91.
- Tarnopolsky M, et al. Can J Neurol Sci 2016;43(4):472–85.
- Kishnani PS, et al. Am J Med Genet A 2013;161(10):2431–43.
- Barohn RJ, et al. Neurol Clin 2014;32(3):569-vii.
- Hereditary myopathy with early respiratory failure. Site Web Genetics Home Reference. Accessible au : https://medlineplus.gov/genetics/condition/hereditary-myopathy-with-early-respiratory-failure/. Consulté le 28 juillet 2020.
- Jones K, et al. Interventions for dysphagia in long-term, progressive muscle disease. Cochrane Database Syst Rev 2016 doi: 10.1002/14651858.CD004303.pub4.
- Mah JK, et al. Neuromuscul Disord 2014;24(6):482–91.
- Gilchrist JM. Semin Respir Crit Care Med 2002;23(3):191–200.
- Ozawa E, et al. Mol Cell Biochem 1999;190:143–51.
- Barnabei MS, et al. Compr Physiol 2011;1(3):1353–63.
- Limb-girdle muscular dystrophy. Site Web Genetics Home Reference. Accessible au : https://medlineplus.gov/genetics/condition/limb-girdle-muscular-dystrophy/#frequency. Consulté le 28 juillet 2020.
- Site Web Limb-girdle Muscular Dystrophies NORD. Accessible au : https://rarediseases.org/rare-diseases/limb-girdle-muscular-dystrophies/. Consulté le 23 avril 2024.
- Siciliano G, et al. Acta Myol 2015;34(1):3–8.
- Myasthenia gravis. Site Web Genetics Home Reference. Accessible au : https://medlineplus.gov/genetics/condition/myasthenia-gravis/. Consulté le 29 juillet 2020.
- Myasthenia gravis: what is it? Site Web de Harvard Health Publishing. Accessible au : https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Consulté le 29 juillet 2020.
- Bernatsky S, et al. Ann Rheum Dis 2009;68:1192–96.
- Polymyositis. Site Web de Johns Hopkins Medicine. Accessible au : https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Consulté le 29 juillet 2020.
- Mastaglia FL, et al. Rheum Dis Clin North Am 2002;28(4):723–41.
- Smoyer-Tomic KE, et al. BMC Musculoskeletal Disord 2012;13:103.
- Furst D, et al. Muscle & Nerve 2012;45:676–83.
- Page d’information sur les myopathies inflammatoires. Site Web du National Institute of Neurological Disorders and Stroke. Accessible au : https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Consultée le 29 juillet 2020.
- Taverna S, et al. Aging (Albany NY) 2020;12(15):15856–74.
- Ng KWP, et al. Front Neurol 2022;13:997551.
- Lierena JC Jr, et al. Arq Neuropsiquiatr 2016;74(2):166–76.
- Barba-Romero MA, et al. Rev Neurol 2012;54(8):497–507.
MAT-CA-2301539F - 12/2024


View-SoloSTAR®-video-(EN)
View-SoloSTAR®-video-(FR)
.jpg/jcr:content/image%20(2).jpg)
View-DoubleSTAR®-video-(FR)
.jpg/jcr:content/image%20(3).jpg)
View-DoubleSTAR®-video-(EN)
.jpg/jcr:content/image%20(1).jpg)