Pompe

By the Time Pompe Disease Slows Them Down, the Damage Has Already Begun2–4
Diagnose and manage Pompe disease early to help your patients stay ahead of their disease2–4
Pompe disease is a life-threatening, progressive neuromuscular disorder that causes irreversible muscle damage1–4
Pompe disease, also referred to as acid maltase deficiency or glycogen storage disease type II (GSDII), is an underrecognized, inherited multisystemic disorder that causes progressive muscle damage, profound disability and premature death.1,4,5
For patients, Pompe disease means progressive respiratory and muscle complications, as well as life-limiting burden.4
Pompe disease is heterogeneous, with varying ages of onset and rates of progression4,6
Prevalence 1 in 40,000 for all forms of Pompe disease1†
IOPD-Infantile-onset Pompe disease 1 in 138,000 people7*
Presentation - Within the first year of life5
Progression - Rapidly progressive and, if left unmanaged, results in death by 2 years of age, most commonly due to cardiorespiratory failure3
LOPD-Late-onset Pompe disease 1 in 57,000 people7*
Presentation - At any point during childhood or adulthood5
Progression - Steadily progressive and can result in premature death, most commonly due to respiratory failure5
Diagnose and manage Pompe disease early to help reduce patient burden4,6,8
*Estimates based on studies from The Netherlands. No prevalence studies have been performed in Canada.
†Based on one study in a New York population.

Pathophysiology behind the Burden of Disease
Pompe disease is caused by a deficiency of acid α-glucosidase (GAA) enzyme activity, resulting in uncontrolled and progressive lysosomal glycogen accumulation in muscle cells throughout the body1,3,4
- In patients that present with Pompe disease before 1 year of age (infantile-onset), GAA enzyme activity may be completely missing; and if some activity remains, it is usually <1% of normal5
Uncontrolled glycogen accumulation causes lysosomes to swell and rupture, resulting in1
- Irreversible cellular damage6,10
- Progressive destruction of skeletal muscle (including respiratory musculature), cardiac muscle and smooth muscle9,10
- Debilitating respiratory, motor, musculoskeletal, cardiac, bulbar, and gastrointestinal (GI) muscular manifestations3,9,13–15
Diagnostic and treatment delays result in unchecked disease progression leading to respiratory complications, muscle damage, life-altering burden and/or death2,16
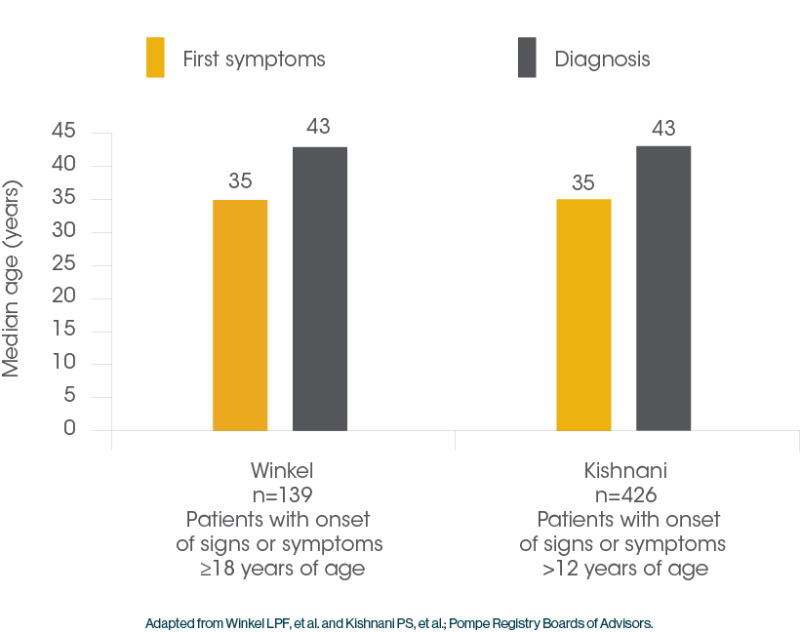
Patients with Pompe Disease often Face Delayed Diagnosis1
-
Delayed diagnosis can lead to significant muscle tissue deterioration, diminished physical function and premature death2,17
- The median delay to definitive diagnosis for patients with LOPD can be as much as 7 years18,19
Deficient GAA enzyme activity leads to progressive muscle damage and multisystemic manifestations1,3-5
Patients with Pompe disease most often present with signs of motor and Respiratory Impairment 4,14,15
- 77% of patients with LOPD present with both exercise intolerance and limb-girdle muscle weakness15
- 55% of patients with LOPD present with both respiratory insufficiency and limb-girdle muscle weakness15
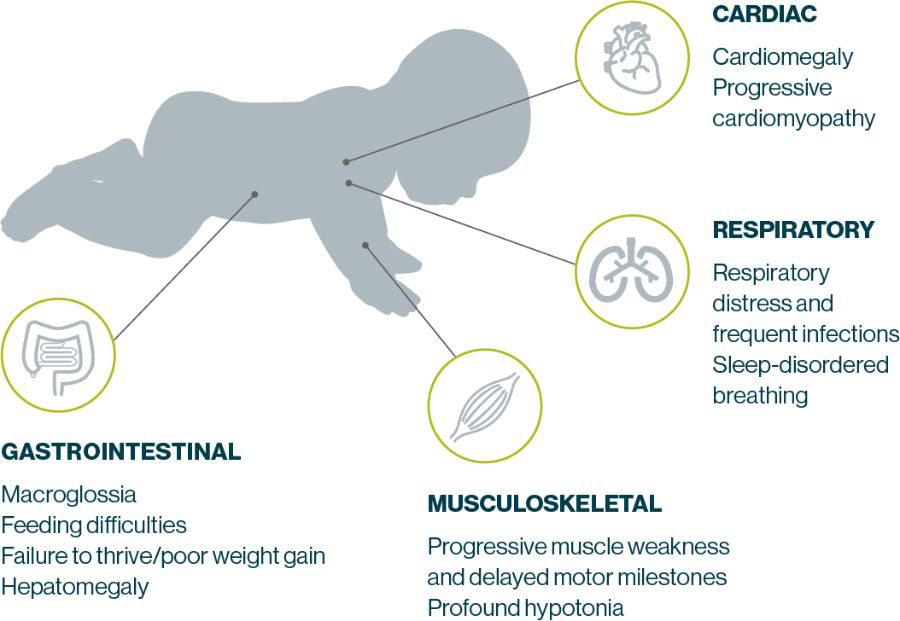
IOPD Symptoms present in early infancy and progress rapidly, typically leading to death by 2 years of age. Manifestations most commonly include cardiomyopathy, hypotonia, muscle weakness, respiratory distress, feeding difficulties, and failure to thrive4,5,20
LOPD can manifest in many different parts of the body
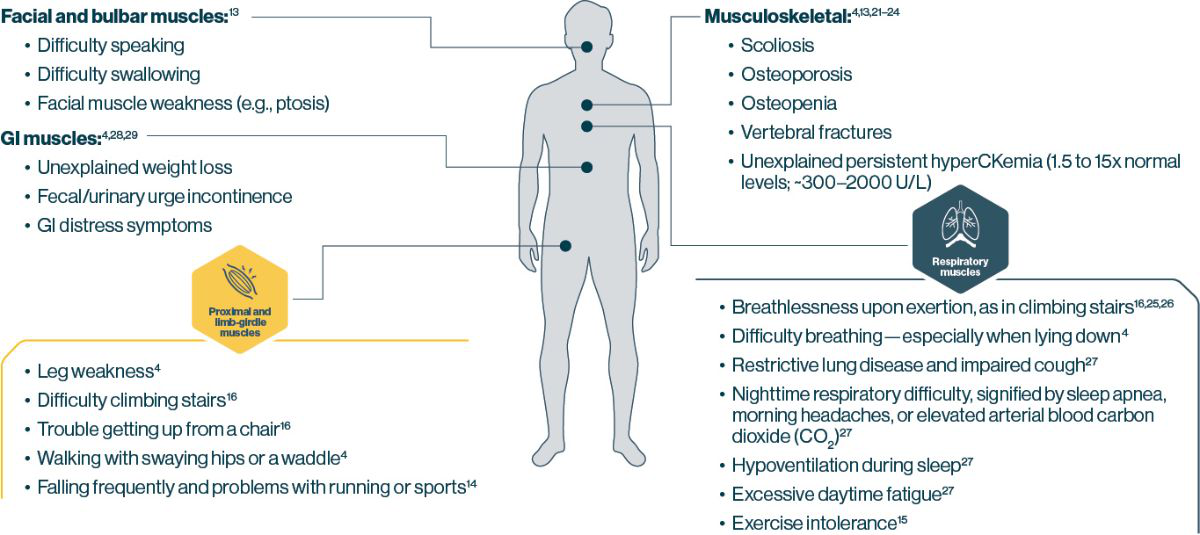
Test early for Pompe disease if you spot these common signs or symptoms in your practice1
Proximal Muscle weakness, the most common presentation in LOPD, makes everyday activities a Challenge for Patients 30

80% of patients with LOPD report experiencing problems with daily tasks due to loss of muscle strength and function22*‡
In patients with LOPD, ongoing and progressive loss of muscle function can lead to life-limiting consequences. At the time of diagnosis:16*
- 50% of patients have difficulty getting up from a chair
- Nearly 50% of patients experience limitations in their ability to work or study
- More than 20% of patients are wheelchair bound and/or require respiratory support
Respiratory Insufficiency is a Cardinal feature of LOPD and the Primary cause of mortality 31
Patients with LOPD who experienced difficulty with tasks at the time of diagnosis 16*†
- Respiratory dysfunction can be the first symptom and may present without overt motor symptoms25
- Symptoms of respiratory insufficiency are the primary presenting symptom in about one-third of LOPD cases25
- Respiratory failure can occur while patients with Pompe disease are still ambulatory4
- Respiratory dysfunction can be present early in the disease course, even when respiratory symptoms are not the initial symptom25,32
- A decrease in FVC is an early sign of respiratory impairment in Pompe disease25
- 4 of 6 patients (67%) showed reduced FVC at diagnosis despite lack of respiratory symptoms32
- Progressive diaphragm weakness is the major cause of respiratory dysfunction in LOPD31
FVC=forced vital capacity.
Modeled progression of respiratory decline – measured by FVC – in untreated patients with LOPD33*

Average respiratory function—measured by FVC—at study entry was 61%, reflecting moderate restrictive lung disease33
- Respiratory function declined consistently, including a 2.3% decline after 12 months and a 6.2% decline after 48 months33
- Respiratory function is often diminished in the early stages of LOPD, prior to diagnosis, and continues to decline over time4,33
The most common cause of death among patients with symptom onset at >12 months of age, reported in the Pompe registry, was respiratory in nature (40.9%) 34
*Based on a meta-analysis of 19 studies of patients with LOPD. Continuous outcomes were modeled using fractional polynomial meta-analysis, which estimates the development of outcomes over time.33
The Differential Diagnosis for Pompe Disease 4
Include Pompe disease in your differential alongside these other diseases
| Muscle weakness | Abnormal gait or difficulty walking | Elevated creatine kinase (CK) | Respiratory insufficiency | Difficulty swallowing | Exercise intolerance | |
| Pompe disease4 2.5 per 100,000 |
 |
|
|
|
|
|
| General myopathy35–39 |  |
|
|
|
|
|
| Duchenne muscular dystrophy35,40–43 1.7–8.2 per 100,000 |
|
|
|
|
|
|
| Limb-girdle muscular dystrophy43–46 0.8–6.9 per 100,000 |
|
|
|
|
||
| Myasthenia gravis40,47,48 20 per 100,000 |
|
|
|
|
|
|
| Inflammatory myopathy49–52 14–25 per 100,000 |
|
|
|
|
||
| Polymyositis52–54 2.7–70 per 100,000 |
|
|
|
|
|
Consider testing for Pompe disease when you see these high-risk signs3,23,55–57
- Presymptomatic, persistent or unexplained sustained elevation of serum creatine kinase (CK) levels: ranging from 1.5 to 15 times the upper limit of normal (~300–2000 U/L)
- Muscle weakness at upper and/or lower limbs (limb-girdle muscle weakness, LGMW)
- HyperCKemia+LGMW
The median delay to definitive diagnosis for patients with LOPD can be as much as 7 years18,19
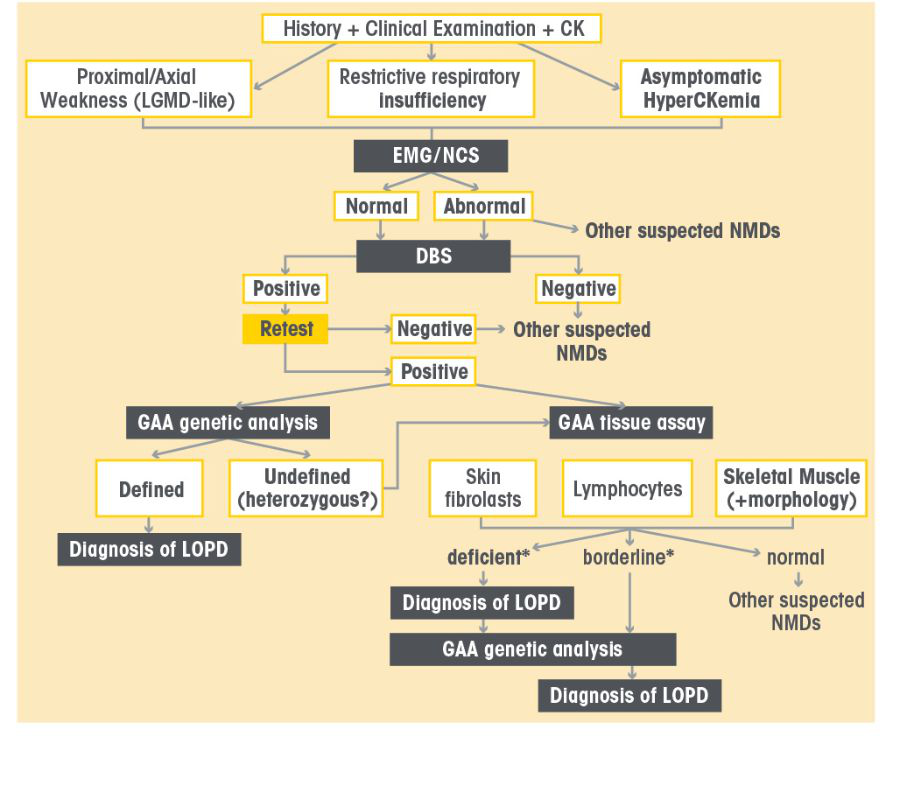
LOPD Diagnostic Algorithm
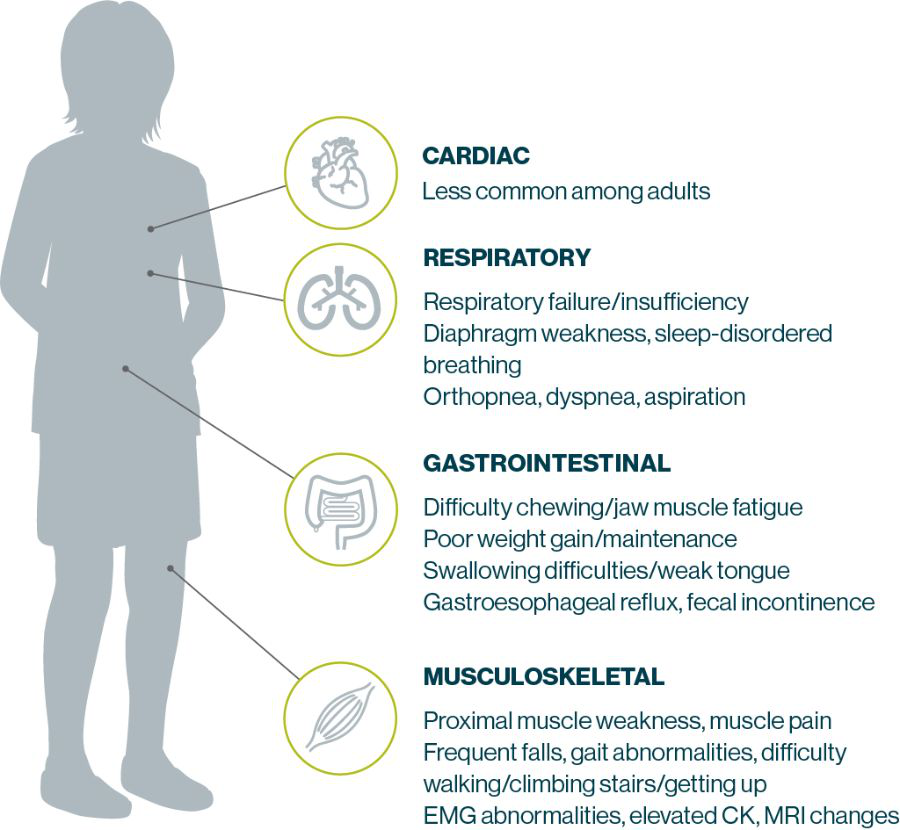
Sign and Symptoms in LOPD Patients
.png)
Sign and Symptoms in IOPD Patients
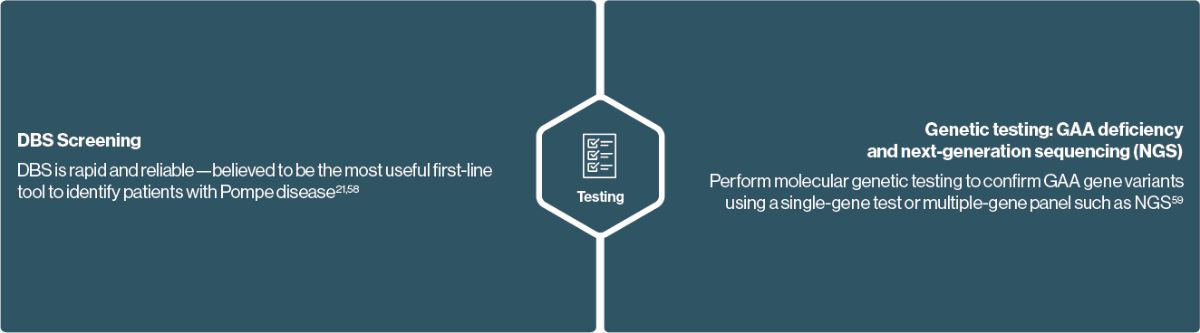
Diagnose Pompe disease using a s imple Dried blood spot (Dbs) Test or Genetic Testing21
GAA enzyme activity testing can identify patients who have Pompe disease21
Testing for Pompe disease
NGS can be used to confirm or test for a number of neuromuscular diseases.
Neuromuscular specialists may have access to NGS to identify the genetic variants involved in the clinical manifestations of Pompe disease. Specifically, the SGM panel examines multiple genes simultaneously, shortening time to diagnosis when you have a large differential consisting of muscular dystrophies, myopathies and myasthenia syndromes.
Consider testing if you suspect Pompe disease or other genetic neuromuscular disorders
GAA enzyme activity testing via DBS is a simple and reliable way to screen for Pompe disease in your symptomatic patients 21

Patient case: 39-year-old woman with progressive respiratory insufficiency 60
Patient presented with a 3-year history of progressive shortness of breath*
Characteristics:
- Progressive shortness of breath
- Shortness of breath upon exertion or within minutes of lying down
- Excessive breathlessness when swimming
- Hypersomnolence during the day with associated morning headaches that developed over the last 2 months
Medical history:
- Epworth score: 21 (out of 24)†
- Unprovoked pulmonary emboli 6 years prior
- Pelvic venous congestion syndrome
- Undergoing persistent anticoagulation therapy
In patients with Pompe disease, ongoing muscle damage can lead to progressive respiratory insufficiency and a variety of other debilitating and potentially life-threatening symptoms1,4,5

Clinical assessment:
- Lab results
- Oxygen saturation on room air: 97%
- Full blood count: normal
- C-reactive protein: normal
- CK: mildly elevated (624 U/L)
- Mobility assessment
- Mildly waddling gait
- Limited weakness on hip flexion
- Pulmonary function test results
- FVC sitting: 2.29 L; 82% predicted
- FVC supine: 0.92 L; 33% predicted
- Daytime arterial blood gas evaluation
- Partial pressure of CO2: 54 mm Hg (upper limit of normal: 45 mm Hg)63
- Partial pressure of oxygen: 85 mm Hg (normal range: 75 to 100 mm Hg)64
- Overnight polysomnogram
- Severe desaturation during rapid eye movement sleep (nadir 68% room air)
Diagnostic screening results:
- Blood spot testing—acid alpha-glucosidase enzyme level activity: <0.1 L (normal range, 0.3-3.0)
Therapeutic interventions:
- Bilevel ventilation on spontaneous-timed mode
- Awaiting start of disease-specific therapy
F/V ex=expiratory portion of flow volume loop; F/V in=inspiratory portion of flow volume loop.
*Not actual patient.
†The Epworth sleepiness scale measures daytime sleepiness via 8 questions on a 4-point scale, from 0 (“would never doze”) to 3 (“high chance of dozing”), that assesses a patient’s propensity to doze or fall asleep during common daily activities.61
Ongoing Management Goals in Pompe Disease
LOPD and IOPD
- Stabilize or improve motor function and muscle strength4,66
- LOPD: ability to stand, sit, walk, and climb stairs
- IOPD: ability to crawl, sit up, and feed
- Stabilize or improve respiratory function65,66
- FVC in upright and supine positions4,9
- Delay the need for respiratory assistance65,66
LOPD
- Delay the need for walking assistance2,65,66
- Maintain or improve quality of life65,66
- Enhance patient independence65,66
IOPD
- Stabilize or improve cardiac health4
- Promote motor development and growth4
In patients with Pompe disease, early and ongoing multidisciplinary care is key to managing symptoms and improving outcomes1,4
- American Association of Neuromuscular & Electrodiagnostic Medicine. Muscle Nerve 2009;40(1):149–60.
- Hagemans MLC, et al. Neurology 2005;64(12):2139–41.
- Hirschhorn R, et al. In: Scriver CR et al., eds. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389–420.
- Kishnani PS, et al. Genet Med 2006;8(5):267–88.
- Kishnani PS, et al. J Pediatr 2004;144:S35–S43.
- Chien YH, et al. J Pediatr 2011;158(6):1023–7.
- Tarnopolsky M, et al. Can J Neurol 2016;43:472–85.
- Alejaldre A, et al. Neuromuscul Disord 2012;22(suppl 2):S148–154. doi:10.1016/j.nmd.2012.05.011.
- Cupler EJ, et al; Muscle Nerve 2012;45(3):319–33.
- Al Jasmi F, et al. BMC Neurology 2015;15:205.
- Thurberg BL, et al. Lab Invest 2006;86(12):1208–20.
- Raben N, et al. Mol Genet Metab 2010;101(4):324–31.
- Chan J, et al. Mol Genet Metab 2017;120(3):163–72.
- van Capelle CI, et al. Orphanet J Rare Dis 2016;11(1):65.
- Schüller A, et al. Am J Med Genet C Semin Med Genet 2012;160C(1):80–8.
- Rigter T, et al. Mol Genet Metab 2012;107(3):448–55.
- Güngör D, et al. Orphanet J Rare Dis 2013;8:49.
- Winkel LPF, et al. J Neurol 2005;252(8):875–84.
- Kishnani PS, et al. Am J Med Genet A 2013;161(10):2431–43.
- Kishnani PS, et al. J Pediatr 2006;148(5):671–6.
- Toscano A, et al. Acta Myol 2013;32(2): 78–81.
- Preisler N, et al. Mol Genet Metab 2013;110(3):287–9.
- Manganelli F, et al. Acta Myol 2013;32(2):82–4.
- Moghadam-Kia S, et al. Cleve Clin J Med 2016;83(1):37–42.
- Mellies U, et al. Respir Med 2009;103(4):477–84.
- Boentert M, et al. Int J Mol Sci 2016;17(10):1735.
- Fuller DD, et al. Respir Physiol Neurobiol 2013;189(2):241–49.
- Karabul N, et al. JIMD Rep 2014;17:53–61.
- Bernstein DL, et al. Mol Genet Metab 2010;101(2-3):130–3.
- Wokke JHJ, et al. Muscle Nerve 2008;38(4):1236–45.
- Wens SC, et al. BMC Pulm Med 2015;15:54.
- Sixel BS, et al. J Bras Pneumol 2017;43(1):54–9.
- Schoser B, et al. J Neurol 2017;264(4):621–30.
- Byrne BJ, et al. Mol Genet Metab 2011;103(1):1–11.
- Jaradeh S. GI Motility Online website. Available at: https://www.nature.com/gimo/contents/pt1/full/gimo35.html. Accessed July 28, 2020.
- Chaudhuri A, et al. Lancet 2004;363(9413):978–88.
- Hereditary myopathy with early respiratory failure. Genetics Home Reference website. Available at: https://medlineplus.gov/genetics/condition/hereditary-myopathy-with-early-respiratory-failure/. Accessed July 28, 2020.
- Barohn RJ, et al. Neurol Clin 2014;32(3):569-vii.
- Féasson L, et al. Ann Readapt Med Phys 2006;49(6):289–300.
- Gilchrist JM. Semin Respir Crit Care Med 2002;23(3):191–200.
- Ozawa E, et al. Mol Cell Biochem 1999;190:143–51.
- Mah JK, et al. Neuromuscul Disord 2014;24(6):482–91.
- Barnabei MS, et al. Compr Physiol 2011;1(3):1353–63.
- Limb-girdle muscular dystrophy. Genetics Home Reference website. Available at: https://medlineplus.gov/genetics/condition/limb-girdle-muscular-dystrophy/#frequency. Accessed July 28, 2020.
- Pegoraro E, et al. In: Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1408/. Accessed July 29, 2020.
- Siciliano G, et al. Acta Myol 2015;34(1):3–8.
- Myasthenia gravis. Genetics Home Reference website. Available at: https://medlineplus.gov/genetics/condition/myasthenia-gravis/. Accessed July 29, 2020.
- Myasthenia gravis: what is it? Harvard Health Publishing website. https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Accessed July 29, 2020.
- Smoyer-Tomic KE, et al. BMC Musculoskeletal Disord 2012;13:103.
- Inflammatory myopathies information page. National Institute of Neurological Disorders and Stroke website. Available at: https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Accessed July 29, 2020.
- Furst D, et al. Muscle & Nerve 2012;45:676–83.
- Mastaglia FL, et al. Rheum Dis Clin North Am 2002;28(4):723–41.
- Polymyositis. Johns Hopkins Medicine website. Available at: https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Accessed July 29, 2020.
- Bernatsky S, Joseph L, Pineau CA. Ann Rheum Dis 2009;68:1192–6.
- Musumeci O, et al. J Neurol Neurosurg Psychiatry 2016;87:5–11.
- Lukacs Z, et al. Neurology 2016;87(3):295–8.
- Leslie N, et al. GeneReviews 2017. Available at : http://www.ncbi. nlm.nih.gov/books/NBK1261. Accessed February 10, 2021.
- Wagner M, et al. Neuromuscul Disor 2013;23(1):89–92.
- Behjati S, et al. Arch Dis Child Educ Pract Ed 2013;98(6):236–8.
- O’Callaghan C, et al. Respirol Case Rep 2016;4(5):e00178.
- Johns MW. Sleep 1991;14(6):540–5.
- Wood KL. Merck Manual Professional Version website. Available at: https://www.merckmanuals.com/professional/pulmonary-disorders/tests-of-pulmonary-function-pft/airflow,-lung-volumes,-and-flow-volume-loop. Accessed July 29, 2020.
- Messina Z, et al. StatPearls. Treasure Island, FL: StatPearls Publishing; 2020. Available at: https://www.ncbi.nlm.nih.gov/books/NBK551648/. Updated November 21, 2019. Accessed July 29, 2020.
- Ortiz-Prado E, et al. Am J Blood Res 2019;9(1):1–14.
- Barba-Romero MA, Barrot E, et al. Rev Neurol 2012;54(8):497–507.
- Llerena JC Jr, et al. Arq Neuropsiquiatr 2016;74(2):166–76.
Diagnose and Manage Pompe Disease early to help Reduce Patient Burden
- In patients with Pompe disease, symptoms of motor impairment are often most pronounced while early respiratory manifestations of diaphragmatic muscle weakness may present with more subtle signs9,14,15
- The leading causes of death are cardiorespiratory failure (IOPD) and respiratory failure (LOPD)4,5,18
- Rapid and definitive diagnostic testing can be done through a DBS GAA enzyme assay combined with a genetic analysis such as NGS21—consider testing for Pompe disease when you see:
- Progressive proximal muscle weakness with or without respiratory insufficiency or unexplained persistent hyperCKemia (1.5 to 15x normal levels; ~300–2000 U/L)1,21-24
- Unspecified limb-girdle myopathy22
- A family member who has Pompe disease5
Connect with Sanofi Genzyme associates for help facilitating discussions around implementing testing protocols for your practice, or for more information, by calling medical information at 1-800-589-6215.

.png/jcr:content/Asset%201%20(2).png)
Prevalence Of LOPD In Patients With Undifferentiated Proximal Myopathy And Undiagnosed Muscle Biopsy
.png/jcr:content/Asset%2010%20(1).png)