
What are LSDs?
Lysosomal storage disorders (LSDs) are rare genetic conditions that cause a buildup of toxic materials in your body’s cells due to enzyme deficiency. LSDs can affect organs such as the brain, spleen, liver, heart, bones, and muscles, which varies depending on the specific disorder.1
While there is no cure for LSDs, early diagnosis is essential to both maximize potential improvement and prevent irreversible organ damage.2

What causes LSDs?
Enzymes are a type of protein that break down certain fats or sugars and assists your cells’ lysosomes with metabolism. Genetic pathogenic variants in the gene encoding an enzyme cause most LSDs. This pathogenic variant leads to accumulation of fats and sugars in cell lysosomes, which disrupts normal function and can cause damage throughout the body.1,3

How are LSDs diagnosed?
Healthcare providers can diagnose LSDs during pregnancy or in children and adults using several diagnostic tests, including enzyme testing and genetic testing.4
What treatments are available?
Treatment of LSDs varies depending on the substances that accumulate. There is currently no cure for LSDs, but several therapeutic options exist including enzyme replacement therapy (ERT), substrate reduction therapy (SRT), chaperone therapy, and hematopoietic stem cell transplantation (HSCT).
TYPES OF LSDs
WORLDWIDE INCIDENCE OF LSDS
1 in 1,500 to 7,000 live births
ACROSS >50 DIFFERENT DISEASES2

Inherited
The majority of LSDs are inherited through an autosomal recessive manner. The three exceptions are X-linked: Fabry disease, Hunter syndrome, and Danon disease.2
SELECT EXAMPLES OF LSDs
| DISEASE | MISSING ENZYME | ACCUMULATING SUBSTANCE |
| ASMD | acid sphingomyelinase | sphingomyelin |
| Fabry disease | α-galactosidase A | globotriaosylceramide (GL-3) |
| Gaucher disease | glucocerebrosidase | glucocerebroside |
| GM2 gangliosidoses | β-hexosaminidase | GM2 ganglioside |
| MPS I | α-L- iduronidase | glycosaminoglycans (GAGs) |
| MPS II | iduronate-2-sulfatase | glycosaminoglycans |
| Pompe disease | acid α-glucosidase | glycogen |
- Demczko M. Merck Manual Professional. 2018. [Epub].
- Staretz-Chacham O et al. Pediatrics. 2009;123(4):1191-1207.
- Griffiths AJF et al. Modern Genetic Analysis. New York: W. H. Freeman; 1999. The Molecular Basis of Mutation. Available from: https://www.ncbi.nlm.nih.gov/ books/NBK21322/. Accessed July 2019.
- Platt, F.M., et al. Nat Rev Dis Primers. 2018; 4, 27.
UNDERSTANDING
ASMD
Historically known as Niemann-Pick disease types A, A/B, and B, acid sphingomyelinase deficiency (ASMD) is a potentially lifethreatening LSD that affects pediatric and adult patients. Deficiency in acid sphingomyelinase (ASM) enzyme activity leads to intracellular sphingomyelin accumulation that results in progressive multiorgan damage. ASMD represents a wide clinical spectrum of disease.1
Anna,
Living with ASMD disease,
Brazil

GENETIC PROFILE
ASMD is an autosomal recessive LSD caused by pathogenic variants in the ASM enzyme–encoding gene sphingomyelin phosphodiesterase 1 (SMPD1).
Sphingomyelin helps regulate cellular processes, such as cell cycle regulation, cell signaling, and apoptosis.
When ASM activity is deficient, sphingomyelin cannot be sufficiently metabolized; it accumulates in lysosomes, particularly in macrophages and hepatocytes. This accumulation damages cells in multiple organs, leading to potentially life-threatening complications.
Males and females have an equal chance of being affected4
Type A
Historically known as Niemann-Pick disease type A (NPA) or infantile neurovisceral ASMD

- Onset: Early Infancy
- Phenotype: Rapidly progressive with acute multiorgan manifestations and neurodegeneration
- Life Expectancy: 2 to 3 years of age
Type A/B
Historically known as Niemann- Pick disease type A/B (NPA/B) or chronic neurovisceral ASMD

- Onset: Infancy to childhood
- Phenotype: Variably progressive with multiorgan manifestations and differing degrees of neurologic involvement
- Life Expectancy: Between early childhood and adulthood
Type B
Historically known as Niemann- Pick disease type B (NPB) or chronic visceral ASMD

- Onset: Infancy to childhood
- Phenotype: Chronic with multiorgan manifestations and no or only minor neurologic involvement
- Life Expectancy: Between childhood and late adulthood
IMPORTANCE OF EARLY DIAGNOSIS
The rarity of ASMD, heterogeneity of its manifestations, and challenging differential diagnosis can result in delayed diagnosis and management of patients.
Early diagnosis of ASMD is critical for appropriate management.

Identifying known disease-causing alleles of ASMD allows for
- Individual genetic counseling
- Carrier screening for at-risk individuals
- Family planning

 |
HOW TO DIAGNOSE |
A simple enzyme activity test can be performed on a DBS (dried blood spot) to indicate reduced ASM activity.

A positive DBS enzyme assay is highly indicative of ASMD, but an analysis from a whole blood sample and/or genetic testing is required to confirm the diagnosis.5
- Wasserstein M et al. Pediatrics. 2004;114;e672.
- Kingma SD et al. Best Pract Res Clin Endocrinol Metab. 2015 Mar;29(2):145-57.
- Wasserstein MP and Schuchman EH. Acid Sphingomyelinase Deficiency. 2006 Dec 7 [Updated 2015 Jun 18]. GeneReviews® [Internet]. Available from: https:// www.ncbi.nlm.nih.gov/books/NBK1370/. Accessed July 2019
- McGovern M et al. Genet Med. 2013;15(8):618-623.
- Wang RY et al. Genet Med. 2011;13(5):457-484.
UNDERSTANDING
FABRY DISEASE
Fabry disease is a rare lysosomal storage disorder in which a deficiency of α-galactosidase A (α -Gal A) causes globotriaosylceramide (GL-3) and other substrates to accumulate in multiple cell types.1,2 Accumulation of GL-3 can lead to progressive cell damage, fibrosis, pain, organ failure, and eventually, premature death if left untreated.3,4
Emma & Signe,
Living with Fabry disease,
Denmark

GENETIC PROFILE
Fabry is inherited in an X-linked manner. This gives females a greater chance of being affected.5 There are over 770 pathogenic variants in the GLA gene that have been reported and they can vary in age of onset, rate of progression, organ involvement, and disease severity.1,15
IMPORTANCE OF EARLY DIAGNOSIS
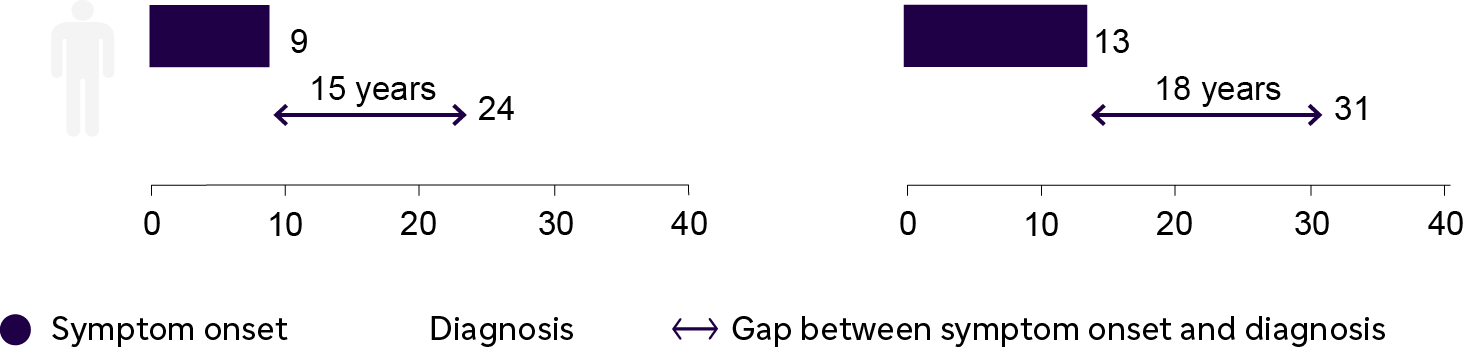
The average time from symptom onset to diagnosis is 15 years for males and 18 years for females.14 The large delay in diagnosis is due in part to the nonspecific nature of early symptoms, heterogeneous phenotypes, and lack of disease awareness.1 Fabry Disease is progressive and can lead to irreversible damage to major body organs. Early diagnosis is very important as it provides an opportunity to intervene early and avoid irreversible organ damage.1
HOW TO DIAGNOSE
An initial diagnosis for Fabry Disease can be made through an α-GAL enzyme assay. This can be an enzyme assay using plasma, leukocytes or skin cultured cells. Another common and easy way to test for α-GAL activity is a DBS (dried blood spot) assay.12
Enzyme assays that show a reduced or absent level of α-GAL should be followed up through a DNA analysis to confirm diagnosis and identify the specific GLA gene mutation. Plasma lyso- GL3 can also be used as a biomarker to confirm diagnosis of Fabry Disease, especially in male patients.17
Genetic testing and counseling should be offered to all relevant family members.13
|
INHERITANCE PATTERN |
||
| X-linked Inheritance | 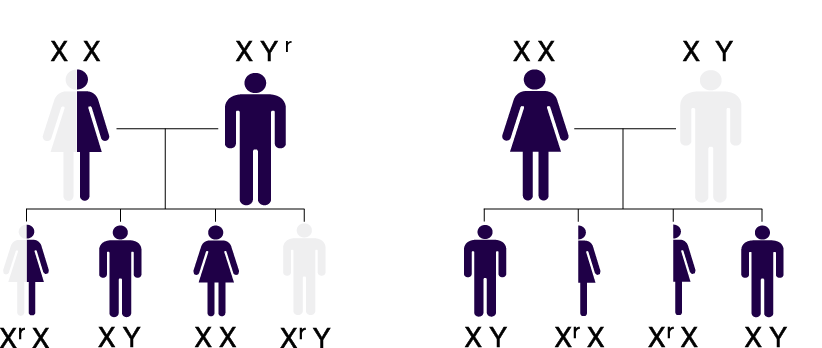 |
Affected mothers have a 50% risk of passing along the defective gene (Xr) to their children, regardless of gender |
 GLA mutation
GLA mutation No GLA mutation
No GLA mutation|
TIME TO DIAGNOSIS |
||
| Male and female patients in the Fabry Registry were diagnosed 15 and 18 years, respectively, after onset of first symptoms1 |
 |
|
- Germain D. Orphanet J Rare Dis. 2010;22;5:30.
- Tsutsumi O et al. Asia Oceania J Obstet Gynaecol. 1985;11:39-45.
- Eng CM et al. Genet Med. 2006;8:539-48.
- Weidemann F et al. Orphanet J Rare Dis. 2013;8:116.
- Desnick RJ et al. α-Galactosidase A Deficiency: Fabry Disease. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62644837. Accessed July 2019.
- Tøndel C et al. J Am Soc Nephrol. 2013;24:137-48.
- Tøndel C et al. Am J Kidney Dis. 2008;51:767-76.
- Schiffmann R and Scott LJC. Acta Paediatr Suppl. 2002;91:48-52.
- Cable WJL et al. Neurology 1982;32:498-502.
- Cole AL et al. J Inherit Metab Dis. 2007;30:943-51.
- Orteu CH et al. Br J Dermatol. 2007;157: 331-7.
- Gal A et al. J Inherit Metab Dis. 2011;34:509-14.
- Laney DA and Fernhoff PM. J Genet Couns. 2008;17:79-83.
- Wilcox WR et al. Mol Genet Metab. 2008;93:112-28.
- Stenson et al (2003), The Human Gene Mutation Database (HGMD®): 2003 Update. Hum Mutat (2003) 21:577-581.
- Poupetova et al. J Inherit Metab Dis. 2010 Aug; 33(4): 387–396.
- Togawa et al. Mol Genet Metab. 2010 Jul;100(3):257-61
UNDERSTANDING
GAUCHER DISEASE
Gaucher disease (pronounced go-shay)
is the most common lysosomal storage
disorder. It is caused by a deficiency in the
enzyme glucocerebrosidase. Insufficient
enzyme activity in Gaucher patients results in
progressive accumulation of glucocerebroside
in the macrophages. Clinical symptoms arise
due to the displacement of normal cells by
lipid engorged Gaucher cells. Accumulation
occurs in organs throughout the body,
typically the bone marrow, liver, and spleen.1,2,3
Pawan,
Living with Gaucher disease,
India

GENETIC PROFILE
Gaucher disease is inherited in an autosomal recessive manner. If both parents are carriers of the diseasecausing allele, their child has a 25% chance of being affected with Gaucher disease. Males and females have an equal chance of being affected.3,6
More than 480 pathogenic variants in the GBA gene are associated with Gaucher disease. N370S [N409S], L444P [L483P], 84GG, and IVS2+1 are the most common pathogenic variants of the GBA gene.8
Homozygosity for L444P (c.1448T>C) is associated with neuronopathic form of the disease and occurs in more than 70% of GD3 patients.8 Worldwide prevalence of neuronopathic Gaucher disease varies substantially by region: East Asia and Middle East 12- 55%, North America and Europe 5%.9-12
IMPORTANCE OF EARLY DIAGNOSIS
Delays in diagnosis of Gaucher disease are common. A patient with Gaucher disease may experience a delay of up to 10 years.7
Gaucher disease is progressive, yet almost 25% of patients do not get timely access to appropriate disease management because of delays in diagnosis.2

|
HOW TO DIAGNOSE |
A simple enzyme activity test can be performed on a DBS (dried blood spot).
A positive DBS enzyme assay is highly indicative of Gaucher disease, but an analysis from a whole blood sample is required to confirm the diagnosis.7
|
PHENOTYPES OF GAUCHER DISEASE PATTERN Gaucher disease is a phenotypic continuum with a spectrum of clinical presentations.13,14 Symptoms can present in early childhood in a severe form or in adulthood in a mild form.4
|

| TYPE 1 Non-neuronopathic |
TYPE 2 Acute neuronopathic |
TYPE 3 Chronic neuronopathic |
|
| NEUROLOGICAL EFFECTS | None | Severe | Moderate to severe |
| SYMPTOM ONSET | Any age | First year of life | Childhood |
| COURSE | Progressive | Rapidly progressive | Progressive |
- Weinreb NJ. Clin Adv Hematol Oncol. 2012;10:3-6.
- Mistry PK, et al. Am J Hematol. 2011;86(1):110-115.
- Grabowski GA et al. Gaucher Disease. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62643884. Accessed July 2019.
- Mehta A. Eur J Intern Med. 2006 Nov; 17 Supply:S2-5.
- Nalysnyk L et al. Hematology. 2017 Mar;22(2):65-73.
- Hopkin RJ and Grabowski GA. Lysosomal Storage Diseases. Harrison’s Principles of Internal Medicine, 20e (http://accessmedicine.mhmedical.com. proxy.libraries.rutgers.edu/content.aspx?bookid=2129§ionid=192531339.) Accessed July 2019.
- Mistry PK et al. Am. J. Hematol. 2007; 82(8): 697-701.
- Institute of Medical Genetics in Cardiff. Human Gene Mutation Database (HGMD) website. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GBA. Accessed October 11, 2022.
- El–Morsy Z, et al. World J Pediatr. 2011;7(4):326–330.
- Abdelwahab M, et al. Neurol Genet. 2016;2(2):e55.
- Tajima A, et al. Mol Genet Metab. 2009;97(4):272–277.
- Choy FY, et al. Blood Cells Mol Dis. 2007;38(3):287–293.
- Grabowski GA, et al. Am J Hematol. 2015;90(suppl 1):S12-S8.
- Goker-Alpan O, et al. J Pediatr. 2003;143(2):273-276.
UNDERSTANDING
GM2 GANGLIOSIDOSES
GM2 gangliosidoses comprise rare, neurodegenerative lysosomal storage disorders, primarily Tay-Sachs disease and Sandhoff disease, caused by deficiency of β-hexosaminidase.
Accumulation of GM2 occurs primarily within neurons and progressively destroys nerve cells in the brain and spinal cord, leading to neurodegenerative symptoms.1,2,3
Zoe,
Living with GM2 gangliosidoses,
USA
Late-onset forms of the disease include debilitating and progressive proximal muscle weakness and imbalance.18

|
Clinical subtypes of GM2 gangliosidoses10,13-15 Subtypes of GM2 gangliosidoses are categorized according to age at onset. |
||
|
Infantile form is aggressive and leads to premature death at approximately 4 years of age |
Death is likely mid-to late-teens |
Variable clinical course and life expectancy |



GENETIC PROFILE
GM2 gangliosidoses are inherited in an autosomal recessive manner. If both parents are carriers of the disease-causing allele, their child has a 25% chance of being affected.
Males and females have an equal chance of being affected.17
IMPORTANCE OF EARLY DIAGNOSIS
The average time from symptom onset to diagnosis for late-onset GM2 gangliosidoses is 16 years. The large delay in diagnosis is due in part to the nonspecific nature of early symptoms and lack of disease awareness. GM2 gangliosidoses are progressive and can lead to irreversible damage to major body organs.18
|
HOW TO DIAGNOSE |
Diagnosis of GM2 gangliosidoses can be accomplished by measurement of Hex A or Hex B enzyme activity.
Confirmatory diagnosis is recommended by detection of two pathogenic variants in HEXA or HEXB .16
|
INCIDENCE AND HIGH-RISK POPULATIONS Incidence is likely to be underestimated due to misdiagnosis and delayed diagnosis resulting from limited disease awareness.4-12 |
 |
 |
||
| Incidence | Carrier frequency | High risk populations | |
| Tay-Sachs disease |
|
|
|
| Sandhoff disease |
|
|
- Neudorfer O et al. Genet Med. 2005;7:119.
- Regier DS et al. Pediatr Endocrinol Rev. 2016;13:663.
- Leal AF et al. Int J Mol Sci. 2020;21:6213.
- Triggs-Raine BL et al. New Engl J Med. 1990;323:6.
- Triggs-Raine BL et al. Am J Hum Genet. 1995;56:870.
- Vallance H and Ford J. Crit Rev Clin Lab Sci. 2003;40:473.
- Frey LC et al. JAMA Neurol. 2005;62:989.
- Neudorfer O et al. Genet Med. 2005;7:119.
- Maegawa GHB et al. Pediatrics. 2006;118:e1550.
- Bley AE et al. Pediatrics. 2011;128:e1233.
- Kaback MM and Desnick RJ. Gene Reviews: Hexosaminidase A deficiency. 2011.
- Lew RM et al. Appl Clin Genet. 2015;8:19.
- Jeyakumar M et al. Neuropath Appl Neurobiol. 2002;28:343.
- Bley AE et al. Pediatrics. 2011;128:e1233.
- Kaback MM and Desnick RJ. Gene Reviews: Hexosaminidase A deficiency. 2011.
- Leal AF et al. Int J Mol Sci. 2020;21:6213.
- Cachon-Gonzalez MB, et al. Curr Gene Ther. 2018;18(2):68-89.
- Toro C et al. Neurosci Lett. 2021 Nov 1;764:136195.
UNDERSTANDING
MPS I
The mucopolysaccharidoses (MPS) are a group of rare, genetic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down storage molecules called glycosaminoglycans
(GAGs).1
MPS I, one of the seven types of MPS, results from deficiency of α-L-iduronidase (IDUA) due to pathogenic variants in the
IDUA gene.1
Kali,
Living with MPS I,
USA

GENETIC PROFILE
MPS I is an autosomal recessive disorder; over 250 pathogenic variants have been described in the literature.1,4
Genotype - phenotype correlations have been established for some pathogenic variants, but others are novel.5
There is a higher prevalence of severe MPS I in the Irish Traveller Community, due to homozygous W402X pathogenic variants.6
IMPORTANCE OF EARLY DIAGNOSIS
Diagnosis of MPS I is often delayed, due to the non-specific nature of symptoms.9 Early diagnosis and intervention is essential to prevent or minimize irreversible organ damage an improve long-term clinical outcomes.10
Diagnosis of Family screening can help siblings of those with MPS I receive an accurate diagnosis and early management.11 MPS I newborn screening has gained increasing importance as the evidence base for early intervention and improved outcomes has been clearly demonstrated.8

 |
SEVERE → ATTENUATED |
MPS I presents as a disease spectrum, ranging from the severe phenotype to the attenuated phenotype2 |
|
HOW TO DIAGNOSE |
A simple urine test to look for abnormally high levels of GAGs (heparan sulfate and dermatan sulfate) can be ordered.1
Diagnosis is confirmed via enzyme assay and genetic testing.2,9
|
INHERITANCE PATTERN |
||
|
Autosomal Recessive Inheritance
|
 |
|

| FOR MORE INFORMATION | https://www.mps1disease.com/ https://mpssociety.org/ https://www.mpsreference.eu/ |
- Neufeld EF and Muenzer J. The Mucopolysaccharidoses. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62642135. Accessed July 2019.
- Muenzer J et al. Pediatrics. 2009;123:19-29.
- Beck M et al. Genet Med. 2014;16(10):759-65.
- The Human Gene Mutation Database. http://www.hgmd.cf.ac.uk/ac/gene. php?gene=IDUA. Accessed July 2019.
- Ghosh A et al. Human Mutation. 2017;38:1555-68.
- Murphy AM et al. Arch Dis Child. 2009;94(1):52-4.
- Bruni S and Lavery C. Mol Genet Metab Rep. 2016;8:67-73.
- Holton J and Ireland JT. Inborn Errors of Skin, Hair, and Connective Tissue.1975:39-60.
- Clarke LA and Atherton AM. J Pediatr. 2017;182:363-70.
- Burlina A et al. J Inherit Metab Dis. 2018;41:209-219.
- Lehman et al. Rheumatology. 2011;50(suppl 5):v41-v48.
UNDERSTANDING
MPS II
The mucopolysaccharidoses (MPS) are a group of rare, genetic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down storage molecules called glycosaminoglycans
(GAGs).1
MPS II, also known as Hunter Syndrome, one of the seven types of MPS, results from deficiency of iduronate-2-sulfatase (IDS) due to the pathogenic variants in the IDS gene.1
David,
Living with MPS II,
Australia

GENETIC PROFILE
MPS II is an X-linked recessive disorder that almost exclusively affects males.1
Heterozygous MPS II females (carriers) are generally asymptomatic or not heavily affected.1
More than 300 pathogenic variants in the IDS gene have been reported. Genotype – phenotype correlations are difficult to establish, but if established, help to better understand the disease.6
Pathogenic variants that result in complete absence of IDS activity lead to a severe phenotype.1,6
IMPORTANCE OF EARLY DIAGNOSIS
Diagnosis of MPS II is often delayed, due to the non-specific nature of symptoms.7 Early diagnosis and intervention is essential to prevent or minimize irreversible organ damage and improve long-term clinical outcomes.7
MPS II newborn screening has gained increasing importance as the evidence base for early intervention and improved outcomes has been clearly demonstrated.8
 |
SEVERE → ATTENUATED |
MPS II presents as a disease spectrum, ranging from the severe (neuronopathic) phenotype to the attenuated (non-neuronopathic) phenotype.3 |
|
HOW TO DIAGNOSE |
A simple urine test to look for abnormally high levels of GAGs (heparan sulfate and dermatan sulfate) can be ordered.1
Diagnosis is confirmed via enzyme assay and genetic testing.9
|
INHERITANCE PATTERN |
||
|
X-Linked Recessive Inheritance
|
 Affected mothers have a 50% risk of passing along the defective gene (Xr) to their children, regardless of gender |
|

| FOR MORE INFORMATION | https://mpssociety.org/ https://www.mpsreference.eu/ |
- Neufeld EF and Muenzer J. The Mucopolysaccharidoses. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62642135. Accessed July 2019.
- Meikle PJ et al. JAMA. 1999;281:249-54.
- Amartino H et al. Mol Genet Metab Rep. 2014;1:401-6.
- Parini R et al. Mol Genet Metab. 2016;117(4):438-46.
- Stapleton M et al. Expert Opin Orphan Drugs. 207;5(4):295-307.
- Martin R et al. Pediatrics. 2008;121:e377-86.
- Wraith JE et al. Eur J Pediatr. 2008;167:267-77.
- Holton J and Ireland JT. Inborn Errors of Skin, Hair, and Connective Tissue.1975:39-60.
- Scarpa M et al. Orphanet J Rare Dis. 2011,6:72.
- Hampe, C. S., et al. Int J Mol Sc.. 2021. 22(15), 7888.
- Hashimoto A.,et al. Otolaryngol. 2018;2018:1–4.
UNDERSTANDING
INFANTILE-ONSET
POMPE
DISEASE
Pompe disease is a progressive, multi-systemic, debilitating, and often life-threatening neuromuscular disorder. Pompe disease is caused by the absence or deficiency of the enzyme acid alpha-glucosidase (GAA), which is responsible for the breakdown of glycogen inside the cells. Without sufficient GAA enzyme, glycogen accumulates primarily in muscle cells, which leads to progressive loss of muscle function. Infantileonset Pompe disease (IOPD) is rapidly progressing and usually presents within the first months of life. If left untreated, IOPD is often fatal before age 1.1
Arathi,
Living with Pompe disease,
India

GENETIC PROFILE
IOPD is inherited in an autosomal recessive manner. The GAA gene can have a spectrum of variants that lead to differing amounts of GAA enzyme activity.1 Variants that leave little to no GAA enzyme activity (<1% of normal) manifest as the infantile onset form of Pompe disease.2
IMPORTANCE OF EARLY DIAGNOSIS
IOPD is rapidly progressing and early diagnosis can be crucial in providing supporting therapy. Delays in diagnosis of IOPD correlate with respiratory failure, profound motor impairment, and death.6
Newborn screening for Pompe disease can readily identify patients with IOPD. This allows immediate diagnosis and can lead to improving survival and outcomes.7
|
HOW TO DIAGNOSE |  |
A relatively quick, simple, and minimally invasive way to screen for Pompe disease is the DBS (dried blood spot) enzyme assay. A DBS is able to detect low or absent levels of the GAA enzyme.3 Reduced GAA enzyme activity should always be followed with a second blood sample and/ or GAA gene sequencing to confirm diagnosis of Pompe disease.3
|
PATHOGENIC GAA VARIANTS CAN PREDICT CLINICAL PHENOTYPE |
||
|
|
 |
 |

| IOPD | LOPD | |
| AGE OF ONSET | Infancy | Infancy - adulthood |
| PROGRESSION | Rapid | Varying |
| PREDOMINANT SYMPTOMS | Respiratory distress, hypotonia, muscle weakness, cardiomyopathy | Progressive proximal muscle weakness, respiratory deficit |
- Reuser AJ et al. Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content. aspx?bookid=971§ionid=62641992. Accessed July 2019.
- Mutations In Human Acid Alpha-glucosidase [database]. Pompe Center Erasmus MC Rotterdam. Revised May 2016. Available at http://cluster15.erasmusmc.nl/klgn/pompe/ mutations. html?lang=en. Accessed July 2019.
- Kishnani PS et al. Genet Med. 2006;8(5):267-288.
- Marsden D. Genet Med. Feb 2005;7(2):147-150.
- Kishnani PS et al. J Pediatr. 2006;148:671-676.
- Chien YH et al. Pediatrics. 2009; 124:e1116–e1125. Supplementary data, available at http://pediatrics.aappublications.org/content/suppl/2009/12/03/124.6.e1116. DC1/2008-3667_SI.pdf. Accessed July 2019.
- Chien YH et al. J Pediatr. 2015; 166:985-91.
- Ausems MG et al. Eur J Hum Genet. 1999;7:713–716.
UNDERSTANDING
LATE-ONSET
POMPE
DISEASE
Pompe disease is a progressive, multi-systemic, debilitating, and often life-threatening neuromuscular disorder. Pompe disease is caused by the absence or deficiency of the enzyme acid alpha-glucosidase (GAA), which is responsible for the breakdown of glycogen inside the cells. Without sufficient GAA enzyme, glycogen accumulates primarily in muscle cells, which leads to progressive loss of muscle function.1 Late-onset Pompe disease (LOPD) has a less rapid and more variable disease course, where symptoms may begin anywhere from infancy to adulthood.1
Andrea,
Living with Pompe disease,
Denmark

GENETIC PROFILE
LOPD is inherited in an autosomal recessive manner. The GAA gene can have a spectrum of variants that lead to differing amounts of GAA enzyme activity.1 The pathogenic variants that leave low to moderate GAA enzyme activity (1-40% of normal) manifest as the less rapid form of Pompe disease, LOPD.8
IMPORTANCE OF EARLY DIAGNOSIS
If treated early enough, muscle damage from glycogen accumulation can be reversible. However, if left untreated, it can lead to irreversible deterioration of skeletal and respiratory muscle, disability, and premature death. The initiation of an intervention is crucial in preventing further deterioration of respiratory function and muscle, and permanent disability.4
A person with LOPD may go several years before receiving a diagnosis. Newborn screening for Pompe disease can help diagnose patients before irreversible muscle damage occurs.7
|
HOW TO DIAGNOSE |
A relatively quick, simple, and minimally invasive way to screen for Pompe disease is the DBS (dried blood spot) enzyme assay. A DBS is able to detect low or absent levels of the GAA enzyme.9
Reduced GAA enzyme activity should always be followed with a second, blood sample and/ or GAA gene sequencing to confirm diagnosis of Pompe Disease.9
|
GENOTYPE - PHENOTYPE CORRELATION |
||
|
|
|
|
| IOPD | LOPD | |
| AGE OF ONSET | Infancy | Infancy - adulthood |
| PROGRESSION | Rapid | Varying |
| PREDOMINANT SYMPTOMS | Respiratory distress, hypotonia, muscle weakness, cardiomyopathy | Progressive proximal muscle weakness, respiratory deficit |
- Reuser AJ et al. Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. http://ommbid.mhmedical.com/content. aspx?bookid=971§ionid=62641992. Accessed July 2019.
- Mutations In Human Acid Alpha-glucosidase [database]. Pompe Center Erasmus MC Rotterdam. Revised May 2016. Available at http://cluster15.erasmusmc.nl/klgn/pompe/.html?lang=en. Accessed July 2019.
- Toscano A. Acta Myol. 2013;32:78–81.
- Thurberg BL et al. Lab Invest. 2006;86:1208-1220.
- Winkel LPF et al. J Neurol. 2005;252(8):875-884.
- Kishnani PS et al. Am J Med Genet A. 2013;161(10):2431-2443.
- Chien YH et al. J Pediatr. 2015; 166:985-91.
- Yang CC et al. Mol Genet Metab. 2011;104:284–8.
- Kishnani PS et al. Genet Med. May 2006;8(5):267-288.
- Ausems MG et al. Eur J Hum Genet. 1999;7:713–716.
Sanofi Rare Disease Registries Program
The Sanofi Registries program is a multi-center, international, longitudinal, observational program that tracks the natural history and outcomes of people with Gaucher, Fabry, MPS I or Pompe disease.

Each Registry is
specifically centered
around the disease and
collects real-world data.

All patients with a confirmed diagnosis are eligible to participate, regardless of their therapy status and choice of treatment. Patients may be enrolled at any time during the course of their disease, including posthumously with ethics approval.

The Registry program is non-interventional and patients receive standard of care treatment as determined by their physicians.

The Registry collects:
• Retrospective and Prospective Data
• Disease Signs and Symptoms
• Treatment Information
• Clinical Assessments
• Patient Reported Outcomes
• Siblings Linkage
GLOBAL REACH OF SANOFI RARE DISEASE REGISTRIES

Guided by the input from
|
4 |
International Boards of Advisors |
|
1 |
Rare Disease Registries Patient Council |
|
12 |
Regional Boards of Advisors |
Fabry, Gaucher,
MPS I, Pompe
4
Main Registries

8
Sub Registries

More Than
19,000
Patients Enrolled
Presence In Over
68 Countries

Over
900 Participating Sites

Support From More Than
1,280 HCPs
FROM PATIENTS FOR PATIENTS


* ePRO = electronic Patient Reported Outcomes available to sites and via patient accounts where available with IRB/EC approval
** Available in 2023 for Gaucher Registry participants and in development for the other Registries
RARE DISEASE REGISTRIES’ CONTRIBUTIONS

INCREASING
DISEASE KNOWLEDGE

IMPROVING
PATIENT CARE


HEALTHCARE POLICY
FORMULATION
REIMBURSEMENT AND ACCESS TO THERAPY


- Mistry, P.K., et al. Orphanet J Rare Dis. 2022; 17, 362.
Supporting Sustainable Access
Sanofi’s Rare Humanitarian Program is the first humanitarian initiative and longest-running program of its kind for people with lysosomal storage disorders. The program is focused on providing access to free treatments for people who meet the program’s criteria, who otherwise would not have access to such treatments, as well as supporting patient diagnosis, treatment monitoring, patient advocacy and physician education.

Over 30 Years
Our Rare Humanitarian Program was established in 1991 when our first treatment for Gaucher disease was approved by the U.S. FDA. Since then, it has evolved and expanded to support six different lysosomal storage disorder communities across six continents.

A Focused Approach
Our global team partners with the patient community, physicians, and Sanofi affiliates to understand and address the needs of the people we serve.

Ensuring Sustainable Care
Our program serves as a resource when access to treatment is limited; support can last anywhere from several months to a decade or more.
Through this program...
3,550+
people in 100+ countries
have received access to free therapy.
1,050+
people in 70+ countries are currently
receiving access to free therapy.
375+
people have received access to free therapy
for over 10 years.
150+
new people receive access to free
therapy each year.
We believe our responsibility does not end with developing effective therapies. We are committed to ensuring sustainable access to approved treatments for patients with lysosomal storage disorders who meet the program’s criteria, regardless of their ability to pay.
Providing humanitarian support to the rare disease community is a vital element of our mission to improve patients’ lives.
It’s in our DNA.

Our Rare Humanitarian Program often serves as a physician’s first experience in treating a lysosomal storage disorder patient. Outside of the program, Sanofi supports training and education for patient identification, understanding of treatment expectations and monitoring.

Sanofi collaborates with local governments to help establish sustainable healthcare systems to care for the needs of patients with lysosomal storage disorders.

Sanofi works closely with patient organizations to help raise awareness of the unique challenges having a lysosomal storage disorder brings and address their unmet needs.
Supporting the Rare Community
At Sanofi we focus on patients and caregivers to ensure we help to support sustainable access to innovation and address unmet needs of the community. We do this through a focused approach and numerous initiatives all aimed at providing better care for rare.
Community Engagement:
We engage with the community to listen, learn and share important developments in the rare ecosystem.
Co-creation:
We don’t work in silos, instead we co-create solutions via multi-stakeholder collaborations and public private partnerships to advance topics important to the rare disease community.

Advocacy:
We work hand-in-hand to help broaden and strengthen the patient and caregiver voice in their advocacy activities.

Education and Awareness:
We participate and drive awareness and education to further elevate key stakeholders understanding of the rare disease community.


The Hill Family,
Fabry Disease*
United States*
Some members of the family in this picture do not have a diagnosis of Fabry Disease

In Specialty Care, our mission is to help people with debilitating and complex conditions in rare diseases, rare blood disorders, neurology, oncology, and immunology. These conditions are often difficult to diagnose and treat. But we aren’t afraid of challenges. They just push us to work harder, to chase new potential therapies that help patients to live their lives.
MAT-BH-2300747-V1-DEC-2023