ASMD Therapie (Typ A/B & B)
Für medizinische Informationen bitte hier einloggen
ASMD im Überblick
ASMD (saurer Sphingomyelinase-Mangel / saure Sphingomyelinase-Defizienz) ist eine schwere, autosomal rezessiv vererbte Stoffwechselkrankheit aus der Gruppe der lysosomalen Speicherkrankheiten. Sie ist mit einer geschätzten Häufigkeit bei Geburt von 0,4 bis 0,6/100.000 Einwohner sehr selten.1
Die Erkrankung ist progressiv, potentiell lebensbedrohlich und multisystemisch. ASMD kann sowohl bei Kindern als auch bei Erwachsenen auftreten. Die moderne Bezeichnung ASMD ersetzt den bisherigen gebräuchlichen Namen Morbus Niemann-Pick Typ A, A/B und B. Früher wurden drei Erkrankungen unter Niemann-Pick zusammengefasst: Typ A (NPA), Typ B (NPB) und Typ C (NPC). Mittlerweile ist bekannt, dass Typ A und Typ B durch einen Mangel an saurer Sphingomyelinase entstehen. Typ C hat jedoch eine andere Ursache, hier kann der Körper Cholesterin und weitere Fettstoffe nicht richtig abbauen.1
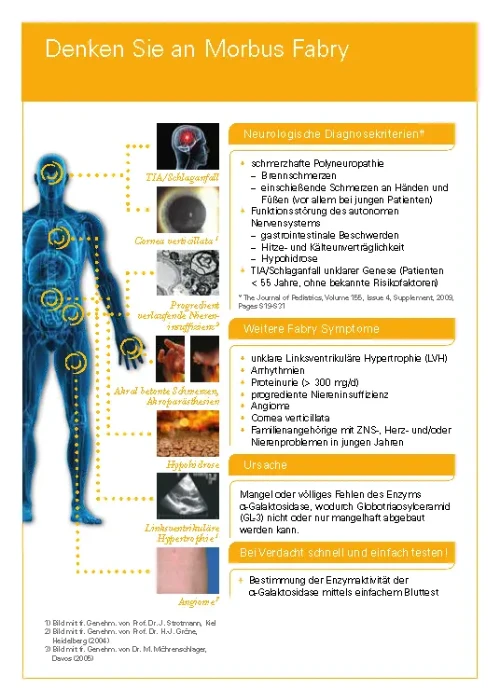
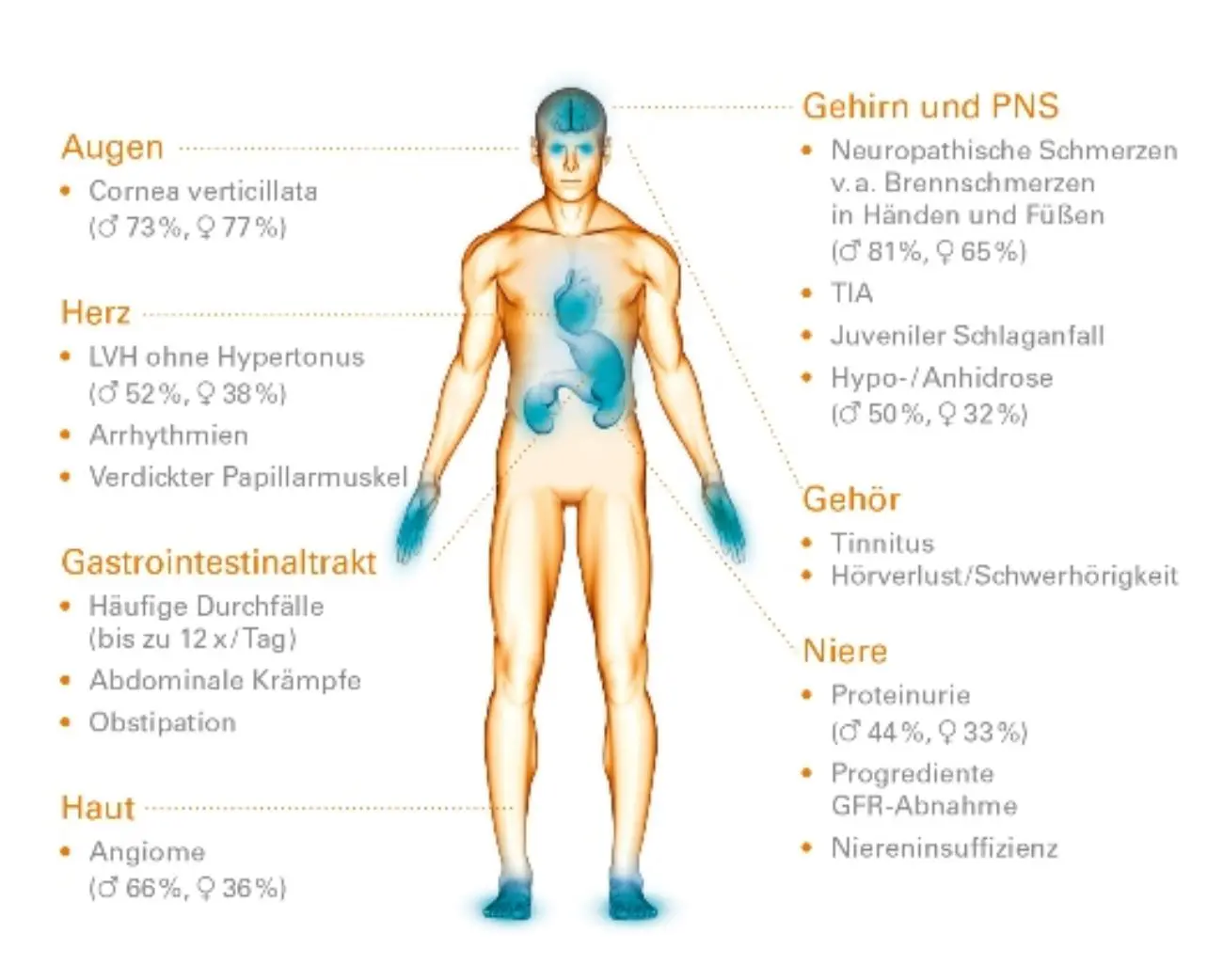
Typische Symptome von ASMD sind eine Vergrößerung der Milz und der Leber, eine interstitielle Lungenerkrankung, eine Verzögerung von Wachstum und Entwicklung sowie eine Verminderung der Anzahl der Blutplättchen (Thrombozyten). Bei einem Teil der Patienten ist auch das zentrale Nervensystem betroffen. Zusätzlich können weitere Organe beteiligt sein, darunter Augen, Herz und Blutgefäße, Knochen und Gelenke sowie das Verdauungssystem. Viele Betroffene berichten von chronischer Müdigkeit.
ASMD weist ein breites Krankheitsspektrum auf, das in drei Subtypen unterteilt wird, wobei die Übergänge fließend sind:
| Typ A | Typ A/B | Typ B
(häufigste Form) |
|
| Verlauf | infantil neuroviszeral | chronisch neuroviszeral | chronisch viszeral |
| Auftreten | kurz nach der Geburt oder in den ersten Lebensmonaten | Säuglingsalter bis Kindheit | kann in jedem Alter auftreten |
| klinische Merkmale | Beeinträchtigung mehrerer Organe, schwerwiegende Störung des zentralen Nervensystems, rasches Fortschreiten der Erkrankung | betrifft wie Typ B mehrere Organe, verschlechtert sich langsamer als Typ A, variable Beteiligung des zentralen Nervensystems | betrifft mehrere Organe, verschlechtert sich langsamer als Typ A, geringe bis keine Beteiligung des zentralen Nervensystems |
| Lebenserwartung | ca. 3 Jahre | zwischen Kindes- und frühem Erwachsenenalter | Erreichen des Erwachsenenalters |
| häufige Todesursachen | Lungenerkrankung, Komplikationen durch Blutungen | Lungenerkrankung, Lebererkrankung | Lungenerkrankung, Lebererkrankung |
Die Diagnose von ASMD ist einerseits eine Herausforderung und andererseits einfach. Die Herausforderung besteht darin, überhaupt eine so seltene Erkrankung in Betracht zu ziehen: Da deutschlandweit nur wenige Menschen an ASMD erkrankt sind, kennen sie viele Ärzte nur aus dem Lehrbuch und sie ist Ihnen im Praxisalltag nicht im Kopf präsent. Mitunter können die Symptome deshalb zunächst fälschlicherweise häufigeren Erkrankungen wie Tumoren, Atemwegserkrankungen oder auch anderen lysosomalen Speicherkrankheiten zugeordnet werden.
Besteht erst einmal der Verdacht auf ASMD, ist die Diagnose einfach: Mit einem einfachen Trockenbluttest lässt sich die Enzymaktivität der sauren Sphingomyelinase im Blut messen. Ist sie erniedrigt, ist die Diagnose damit gesichert. Zusätzlich erfolgt zur Bestätigung meist eine Analyse des SMPD1-Gens, zumal sich bei einigen Varianten der Verlauf voraussagen lässt.
Da die Erkrankung immer weiter fortschreitet, ist eine frühe Diagnose wichtig, um frühzeitig mit der Therapie beginnen zu können.
Für ASMD Typ A/B und Typ B ist eine Behandlungsmöglichkeit in Form einer Enzymersatztherapie verfügbar. Hierbei wird dem Körper eine biotechnologisch hergestellte Version des fehlenden Enzyms ASM regelmäßig per Infusion zugeführt. Ziel ist es, das bereits gespeicherte Sphingomyelin abzubauen und die Ablagerung von weiterem Speichermaterial zu verhindern. Da das infundierte Enzym die Blut-Hirn-Schranke nicht passieren und nicht in das Gehirn gelangen kann, können die derzeitigen Behandlungsmöglichkeiten nur die viszeralen (=die Organe betreffenden), aber nicht die neurologischen Symptome von ASMD verbessern.
- McGovern MM et al. Genet Med 2017; 19: 967-974
Fabry-Mann_Vorteilskarte
.2024-02-07-10-31-33.webp)
1871_DK_Fabry_Neuro_V4_Page_1