aTTP: acquired TTP
Für medizinische Informationen bitte hier einloggen
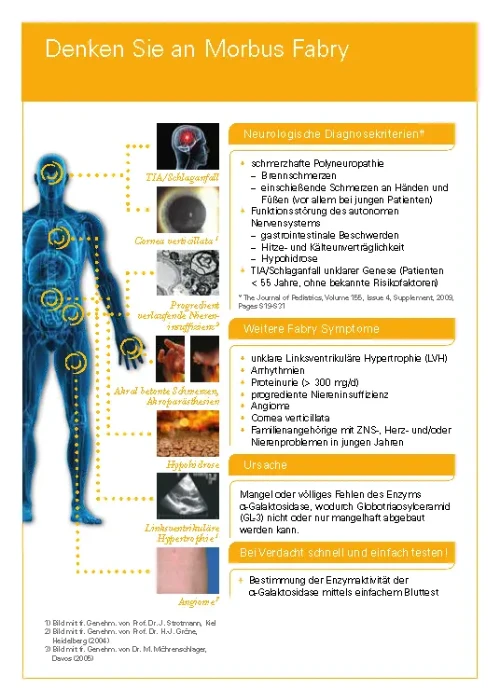
WAS IST TTP?
Die thrombotisch-thrombozytopenische Purpura (TTP) ist eine Form der thrombotischen Mikroangiopathie (TMA). Eine TMA ist in Betracht zu ziehen bei Patienten mit

Die ischämischen Organschäden sind eine Folge der Bildung von Mikrothromben in den kleinsten Gefäßen sowie der Anämie.1,2 Bei der TTP, auch als Moschcowitz Syndrom bekannt (benannt nach dem Erstbeschreiber), sind insbesondere das Gehirn, der Gastrointestinaltrakt, die Niere oder das Herz betroffen.1
Ursache ist die verminderte Aktivität der Protease ADAMTS13. Es wird unterschieden zwischen der erworbenen Form (acquired TTP, aTTP), verursacht durch die Entwicklung von Autoantikörpern gegen ADAMTS13, und der angeborenen Form (congenital TTP, cTTP, auch bekannt als Upshaw-Schulman-Syndrom), die auf Mutationen im ADAMTS13-Gen beruht. Dabei ist die aTTP mit 95 % aller Fälle weitaus häufiger als die cTTP.3
Die Inzidenz der TTP in Europa wird auf 1,5-6/Millionen Menschen pro Jahr geschätzt.4 Ohne Behandlung liegt die Akutletalitätsrate bei bis zu 90 % .1,5 Auch unter der bisher verfügbaren Therapie betrug die Letalität noch bis zu 20 %4. Bis zu 84 % der aTTP Patienten haben ein lebenslanges Rückfallrisiko.18
Die bisherige Therapie der aTTP bestand aus Plasmapherese und Immunsuppression.3,5,6 Seit 2018 ist zusätzlich eine spezifische Therapie verfügbar.3,6,7
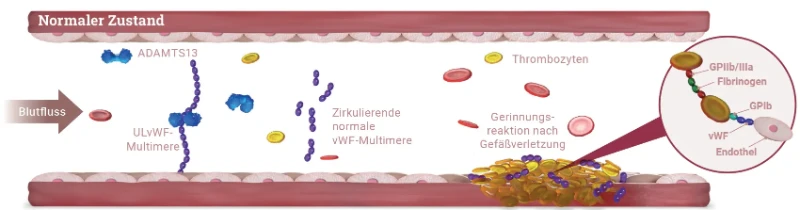
Bei der TTP Erkrankung kommt es zu einer Anreicherung von nicht-prozessierten, ultralangen von-Willebrand-Faktor (vWF)-Multimeren im Blut, an die vermehrt Thrombozyten binden. Dies führt zu
- einer Bildung von disseminierten mikrovaskulären Thromben,
- einer häufig schweren Thrombozytopenie aufgrund des Verbrauchs von Thrombozyten und zu
- einer mikroangiopathischen hämolytischen Anämie (MAHA) durch die mechanische Beschädigung von Erythrozyten, (Schistozyten/Fragementozyten) die auf Scherkräfte in den durch die Mikrothromben verengten kleinsten Gefäße zurückzuführen ist.1,5
Die Blutgerinnsel und die Anämie führen zu ischämischen Organschäden, die abhängig vom beteiligten Kapillarendstromgebiet in verschiedensten Organen auftreten können. Bei der TTP besonders häufig betroffen sind das Gehirn, der Gastrointestinaltrakt, die Niere und das Herz.1
Die Rolle von ADAMTS13 bei der erworbenen TTP
Ursache für die nicht ausreichende Prozessierung der ultralangen vWF-Multimere ist die deutlich verminderte Aktivität der Protease ADAMTS13 (A Disintegrin And Metalloprotease with ThromboSpondin-1 like domain 13), welche die ultralangen Multimere normalerweise spaltet.
Bei der aTTP Krankheit reduzieren Autoantikörper gegen ADAMTS13 die Aktivität des Enzyms. Dagegen tritt die verminderte Aktivität bei der äußerst seltenen cTTP aufgrund von Mutationen im ADAMTS13-Gen auf.1,5
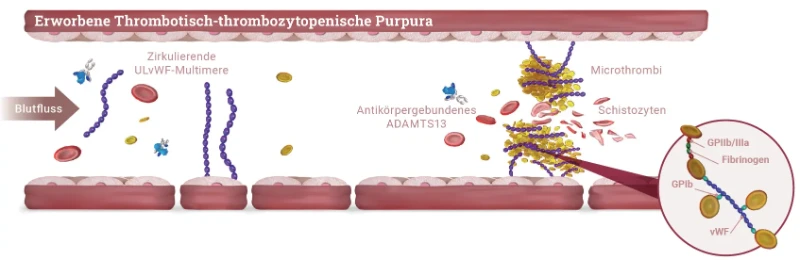
Nach Joly 2017⁹
Der Grund, warum manche Menschen eine aTTP entwickeln, ist nicht bekannt. Verschiedene medizinische Umstände werden in Zusammenhang mit TTP aber immer wieder beobachtet und könnten eine Rolle als mögliche Auslöser spielen:9
- Infektionen
- Autoimmunerkrankungen, besonders systemischer Lupus erythematodes (SLE), Antiphospholipid-Syndrom, Gougerot-Sjögren-Syndrom
- Schwangerschaft
- Medikamente
- HIV-Infektion
- Pankreatitis
- Krebs
- Organtransplantation
Der Beginn einer Episode der thrombotisch thrombozytopenen Purpura ist meist akut und der Krankheitsverlauf schwer bis lebensbedrohlich.1
Die klinische Symptomatik während einer akuten Episode ist Ausdruck einer Organischämie aufgrund der Bildung von disseminierten mikrovaskulären Thromben und einer Anämie. Die Symptome können dabei sehr unspezifisch sein und das Krankheitsbild, abhängig vom vorrangig betroffenem Organsystem, sehr unterschiedlich ausfallen1,5. Aufgrund der meist schweren Thrombozytopenie können zudem Blutungen im Körper oder unter der Haut (Petechien) auftreten.
Übersicht möglicher Symptome2,5,9:

Unbehandelt entwickeln bis zu 90 % der Patienten während einer akuten TTP-Episode ein multiples Organversagen und versterben innerhalb von Tagen bis Wochen nach Beginn der Episode.1
Nach einer ersten Episode bleibt ein Risiko für weitere Schübe bestehen. Als potenzielle Langzeitfolgen können unter anderem Bluthochdruck und kognitive Leistungseinschränkungen auftreten. Zudem leiden Betroffene im Vergleich zur Allgemeinbevölkerung häufiger an Depressionen.4,11
TTP ist ein medizinischer Notfall. Eine schnelle Diagnose und ein früher Therapiebeginn können Leben retten.5 Gemäß internationalen Leitlinien (ISTH) ist eine TTP bei Patienten mit Thrombozytopenie und mikroangiopathischer hämolytischer Anämie bei relativ erhaltener Nierenfunktion in Betracht zu ziehen3:
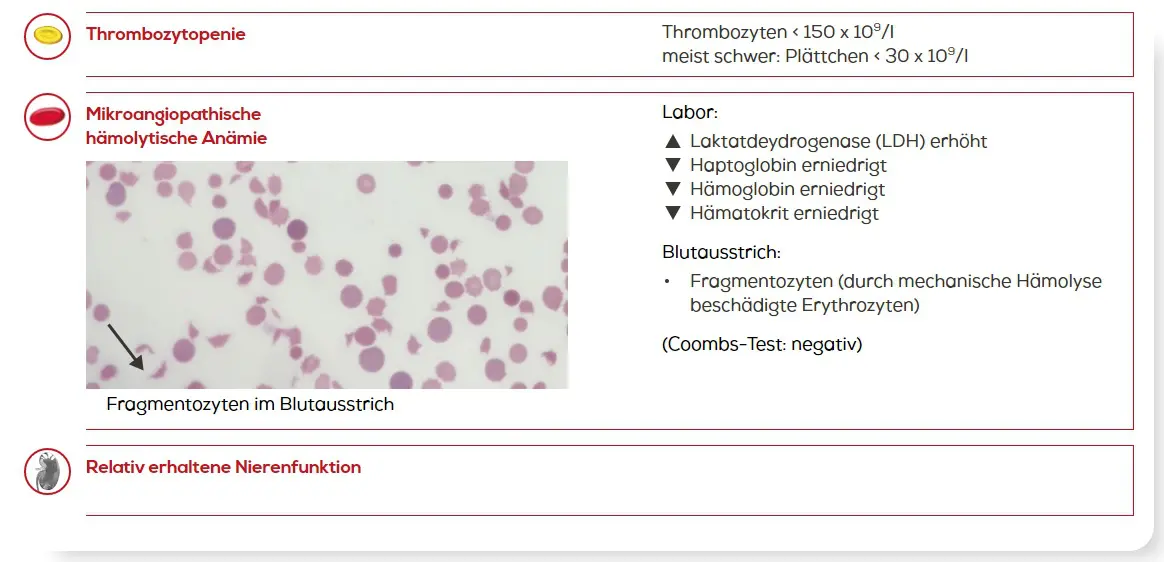
Mögliche Laborbefunde bei TTP2, 3, 5, 9
Neben diesen Laborbefunden und den klinischen Symptomen (s.o.) können bestimmte erhöhte Laborwerte, v.a. LDH, Troponin oder Kreatinin, auf eine Organschädigung hinweisen und so den Verdacht in Richtung TTP lenken.
Differentialdiagnose
Die Differentialdiagnose der aTTP umfasst diverse Formen von thrombotischen Mikroangiopathien. Dazu gehören u. a. das Hämolytisch-Urämische Syndrom (HUS) in seinen unterschiedlichen Ausprägungen (STEC-HUS, aHUS, SP-HUS) sowie verschiedene sekundäre TMA-Formen.2
Hämolytisch-Urämisches Syndrom (HUS)
An ein HUS ist vor allem bei TMA mit einer starken Nierenbeteiligung, bis hin zum akuten Nierenversagen mit Urämie, zu denken. Am häufigsten ist das STEC-HUS, das durch Infektionen mit Shigatoxin produzierenden Escherichia coli Stämen ausgelöst wird. Sehr selten tritt ein HUS nach einer Infektion mit Streptococcus pneumoniae auf (SP-HUS). Liegt keine entsprechende Infektion vor und ist eine schwere ADAMTS13-Defizienz ausgeschlossen, kommt ein atypisches Hämolytisch-Urämisches Syndrom (aHUS) infrage, dem eine übermäßige Aktivierung des Komplementsystems zugrunde liegt und das daher auch als Komplement-vermitteltes HUS bezeichnet wird.2
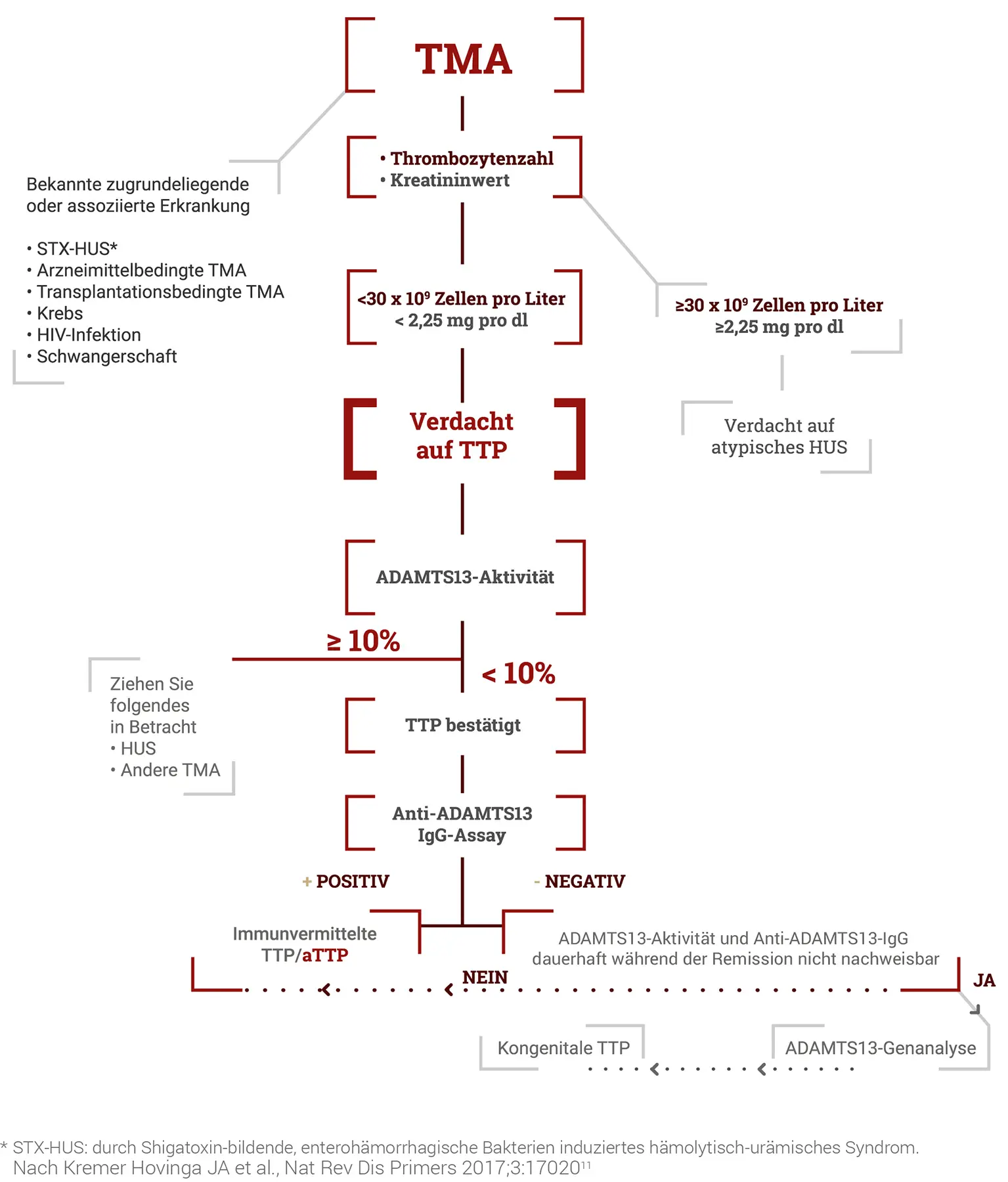
Diagnose und Differentialdiagnose der TTP innerhalb der TMA; nach1
Sekundäre TMA
Eine sekundäre TMA ist in Betracht zu ziehen, wenn
- eine Krebserkrankung,
- eine Infektion, einschl. HIV-Infektion oder
- eine Autoimmunerkrankung,
- eine Schwangerschaft
vorliegt. Zudem können Organ- oder Stammzelltransplantationen und auch bestimmte Arzneimittel (Ticlopidin, Chinin, Mitomycin, Tacrolimus, Gemcitabin, u.a.) eine TMA auslösen.2
ADAMTS13-Testung
Bestätigt wird die Diagnose der TTP durch den Nachweis einer erniedrigten ADAMTS13-Aktivität im Plasma von weniger als 10 % des Normwerts bzw. 10 IE/dl.3 Das Blut für diese (und auch alle anderen) Tests sollte vor Einleitung therapeutischer Maßnahmen, besonders eines Plasmaaustausches, abgenommen werden.3
Bei einer aTTP lassen sich bei der überwiegenden Mehrheit der Patienten Antikörper gegen das Enzym im Blut nachweisen. Bei einer cTTP sind keine Antikörper vorhanden; sie wird über den Nachweis von Mutationen im ADAMTS13-Gen bestätigt.
Der French Score beruht auf einer Analyse des französischen TMA-Registers. Demnach wiesen ein Serum-Kreatinin-Level ≤ 2,26 mg/dl (200 µmol/l) und eine Thrombozytenzahl < 30 x 109/l eine hohe Assoziation mit einer schweren ADAMTS13-Defizienz auf. Durch die Verwendung dieser beiden Kriterien wurden nur sehr wenige Patienten falsch klassifiziert.13
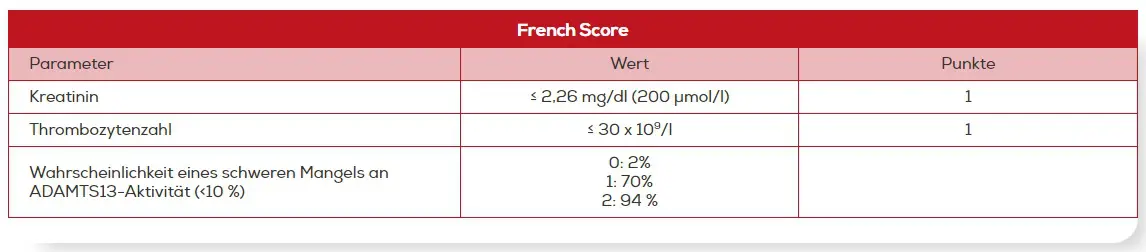
French Score: Abschätzung der Wahrscheinlichkeit für das Vorliegen einer ADAMTS13-Defizienz13
Der PLASMIC Score bezieht insgesamt 7 Kriterien ein. Je mehr dieser Kriterien bei Patienten mit TMA erfüllt werden, desto höher ist die Wahrscheinlichkeit für eine TTP: Mehr als 80 % der Patienten in der Kohorte mit einem Score von 6 oder 7 hatten eine schwere ADAMTS13-Defizienz. Andersherum wiesen die Patienten mit einer bestätigten TTP einen medianen Score von 7 auf (IQR 6-7).14
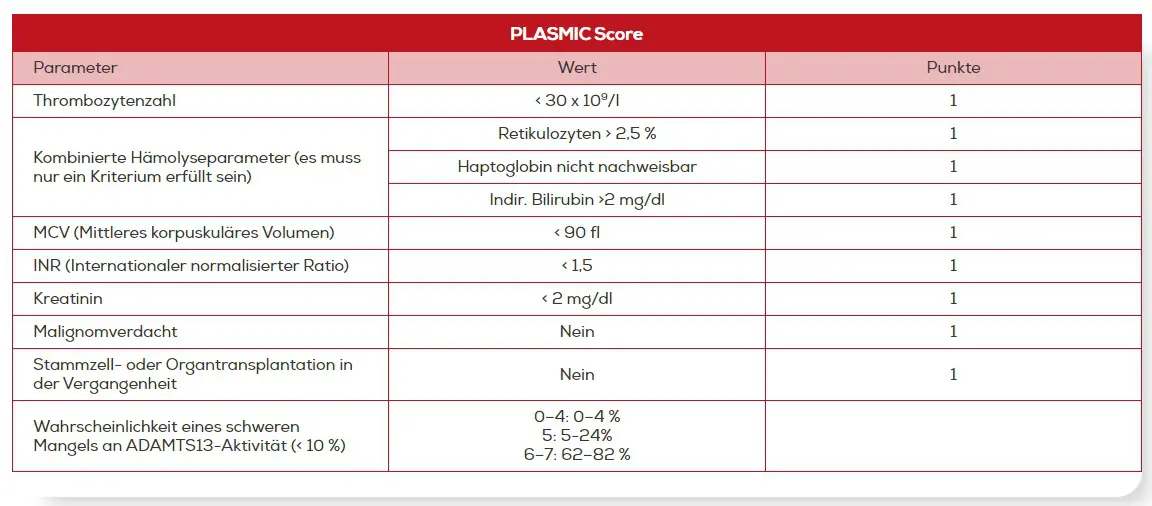
PLASMIC Score zur Abschätzung der Wahrscheinlichkeit für das Vorliegen einer ADAMTS13-Defizienz.14
- Kremer Hovinga JA et al. Nat Rev Dis Primers 2017; 3: 17020
- Bommer M et al. Deutsches Ärzteblatt 2018; 115: 327-334
- Zheng XL et al. J Thromb Haemost 2020; doi:10.1111/JTH.15006 (Diagnosis)
- Miesbach W et al. Orphanet J Rare Dis 2019; 14: 260
- Scully M et al. Br J Haematol 2012; 158: 323-335
- Zheng XL, et al. J Thromb Haemost 2020; doi:10.1111/JTH.15010 (Treatment)
- Fachinformation
- Reese JA et al. Pediatr Blood Cancer 2013;60:1676-1682
- Joly BS et al. Blood 2017; 129: 2836-2846
- Saad J, Schoenberger L. Physiology, Platelet Activation. [Update 27.10.2018]. In: StatPearls [Internet]. Erhältlich unter: https://www.ncbi.nlm.nih.gov/books/NBK482478/ (Letzter Zugriff: 10/2020)
- Vesely SK. J Thromb Haemost 2015; 13: S216-S222
- Sawler D et al. Tromb Res 2020; 193: 53-59
- Coppo P et al. PLoS ONE 2010; 5: e10208
- Bendapudi PK et al. Lancet Haematol 2017; 4: e157-e164
- Jamme M, Rondeau E. Lancet Haematol 2017; 4: e148-e149
- Goel R et al. Transfusion 2016; 56: 1451-1458
- Coppo P, Veyradier A. Presse Med 2012; 41: e163-176
- Thejeel B, et al. Am J Hematol 2016; 91: 623-630
- Sayani FA, Abrams CS. Blood 2015; 125: 3860-386
Fabry-Mann_Vorteilskarte
.2024-02-07-10-31-33.webp)
1871_DK_Fabry_Neuro_V4_Page_1