- Article
- Source: Campus Sanofi
O que é a doença de Fabry?

A doença de Fabry é uma doença rara hereditária, multissistémica e progressiva ligada ao cromossoma X, que pode afetar qualquer indivíduo independentemente da sua idade e origem étnica.
A doença foi descrita pela primeira vez em 1898 pelo Dr. William Anderson (Inglaterra) e Johannes Fabry (Alemanha), de forma independente. A doença de Fabry é também conhecida como angiokeratoma corporis diffusum universal e ainda como doença de Anderson-Fabry.1
Fisiopatologia da doença de Fabry
A doença de Fabry é um dos mais de setenta transtornos raros hereditários designados por doenças lisossomais de sobrecarga. Cada uma destas doenças é desencadeada por uma variante genética patogénica que resulta na deficiência de uma ou mais enzimas lisossomais específicas. A idade com que se iniciam os sintomas, o(s) sistema(s) de órgãos afetados e a gravidade da doença variam significativamente, mas todas elas são progressivas.
A doença de Fabry é causada por uma variante patogénica no gene GLA que codifica a enzima lisossomal α-galactosidase A (também conhecida como α-GAL; α-Gal-A, e ceramida trihexosidase).1 A deficiência parcial ou total na atividade da α-GAL resulta na capacidade reduzida de catabolização de glicoesfingolípidos com resíduos terminais de α-galactosil. Estes glicoesfingolípidos, especialmente as globotriaosilceramidas (designadas por vezes como GL-3 ou Gb-3, ceramida trihexoside), acumulam-se nos lisossomas de vários tipos de células, incluindo as células endoteliais capilares, renais, cardíacas e nervosas, tendo como consequência o dano multissistémico progressivo.
Com o passar do tempo, esta situação pode evoluir até ao dano terminal de determinados órgãos como os rins, coração e sistema cerebrovascular.
Acumulação de GL-3 nas células endoteliais capilares
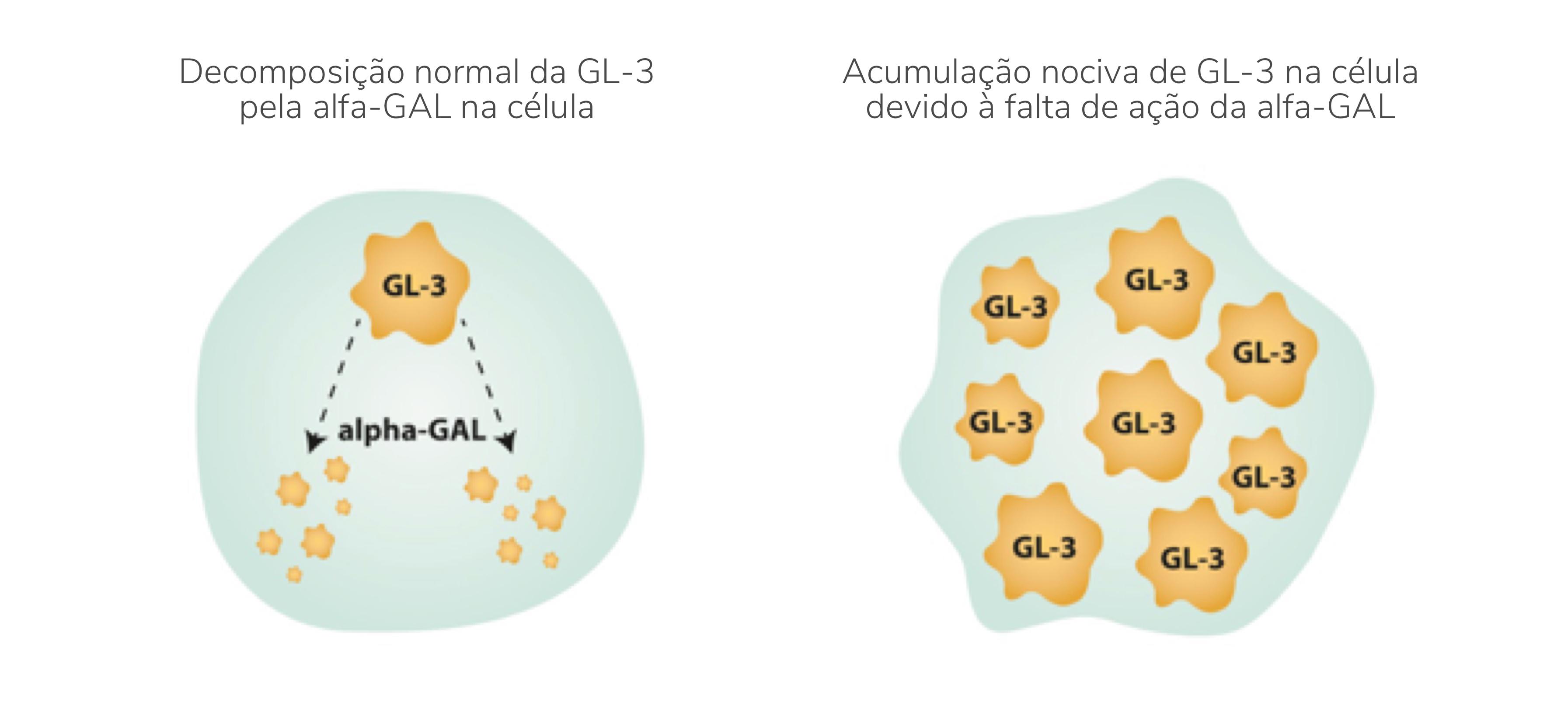
Apresentação da Doença
A doença de Fabry é uma doença multissistémica com um espectro de fenótipos clínicos heterogéneos e progressivos. Têm sido descritas variantes clássicas e não-clássicas (também conhecidas como atípicas ou de início tardio) da doença de Fabry. No fenótipo “clássico” de doença de Fabry, o início dos sinais e sintomas ocorre geralmente ainda durante a infância ou adolescência. Já no que diz respeito ao fenótipo não-clássico, os primeiros sinais e sintomas surgem numa fase mais tardia, variando marcadamente de indivíduo para indivíduo. A grande variedade dos fenótipos clínicos documentados em ambas as formas da doença dificulta o diagnóstico.1,2
Em pessoas com doença de Fabry, a acumulação progressiva de GL-3 inicia-se precocemente e pode prolongar-se ao longo de décadas.1,2 A evolução clínica precoce da doença envolve tipicamente sintomas que afetam primariamente a qualidade de vida, como demonstrado na figura abaixo. A acumulação progressiva de GL-3 em vários tipos de células em todo o organismo pode levar a manifestações potencialmente fatais, envolvendo principalmente os rins, o coração e os sistemas nervoso e cerebrovascular.1 Tipicamente, 50% dos homens e mulheres têm eventos - renais, cardíacos e cerebrovasculares - entre os 40 e os 50 anos de idade, respetivamente. No entanto, verificou-se que, em homens ocorreram eventos antes dos 10 anos de idade e em mulheres após os 20 anos.3 O facto da doença de Fabry possuir um espetro tão alargado e a natureza inespecífica de sinais e sintomas leva a que estes sejam por vezes desvalorizados e/ou atribuídos a outras causas. Isto pode resultar no atraso do diagnóstico, muitas vezes já na presença de danos nos órgãos alvo.1 O diagnóstico precoce facilita a gestão adequada da doença e dos seus sintomas.
A análise dos dados dos primeiros 1765 doentes incluídos no Registo de Fabry mostrou uma tendência para o início dos sintomas entre os 9 e 23 anos nos homens e entre os 13 e 32 nas mulheres, indicando atrasos no diagnóstico em ambos os sexos.4
Um ou mais dos achados listados abaixo podem apontar para a doença de Fabry. Quando na presença de algum destes, a doença de Fabry deve ser incluída no diagnóstico diferencial:
- Angioqueratomas
- Disfunção cardíaca
- Intolerância ao frio e/ou calor
- Hipo-hidrose/anidrose
- Manifestações neurológicas
- Achados oftalmológicos
- Dor
- Manifestações Renais
- Desnick R.J., Ioannou Y.A., Eng C.M. (2014). α-Galactosidase A Deficiency: Fabry Disease. In Valle D, Beaudet A.L., Vogelstein B, Kinzler K.W., Antonarakis S.E., Ballabio A, Gibson K, Mitchell G (Eds), . Retrieved December 11, 2015.
- Germain DP. Fabry Disease. Orphanet J Rare Dis. 2010;5:30.
- Schiffmann R., Warnock DG, Banikazemi M, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009 Jul;24(7):2102-11
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007 Apr; 30(2):184-92
Caminhar

Levantar-se-de-uma-cadeira

Subir-escadas

Levantar-os-braços-acima-da-cabeça

Vídeo-doença-de-Pompe2

Vídeo-Movimentos-de-Pompe---MARCHA_HCPs2

Vídeo-Movimentos-de-Pompe---SUBIR_ESCADAS_HCPs2

Vídeo-Movimentos-de-Pompe---SUBIR_ESCADAS_HCPs3

Vídeo-McIntosh-et-al-20202

Living-with-Pompe-disease-Sean-s-and-Cheryl-s-story

Testemunho-Shaylee-2
