- Article
- Source: Campus Sanofi
Генетика та епідеміологія
Поширеність захворювання
Хоча хвороба Гоше є рідкісним захворюванням, вона є однією з найбільш поширених лізосомальних хвороб накопичення, так само як і хвороба Фабрі.
- Тип 1 (ненейропатичний) є найчастішою формою захворювання, поширеність якої становить 1 на 50 000-100 000 осіб. Хоча хвороба Гоше може виникати в будь-якій з етнічних груп, особливо висока поширеність хвороби Гоше 1 типу спостерігається в окремих етнічних групах, зокрема серед євреїв ашкеназі: поширеність цього захворювання може досягати приблизно 1 на 850 осіб. Вважається, що більш ніж 90% всіх випадків хвороби Гоше становить 1 тип захворювання.1,2
- Хвороба Гоше 2 та 3 типу зустрічається рідко, поширеність цих типів становить <1 на 100 000 осіб.3
- Більш висока поширеність захворювання 3 типу зареєстрована у пацієнтів із провінції Норрботтен в Швеції.4
Як і для будь-якого рідкісного захворювання, точну кількість осіб, які мають хворобу Гоше, визначити складно.
Успадкування хвороби Гоше
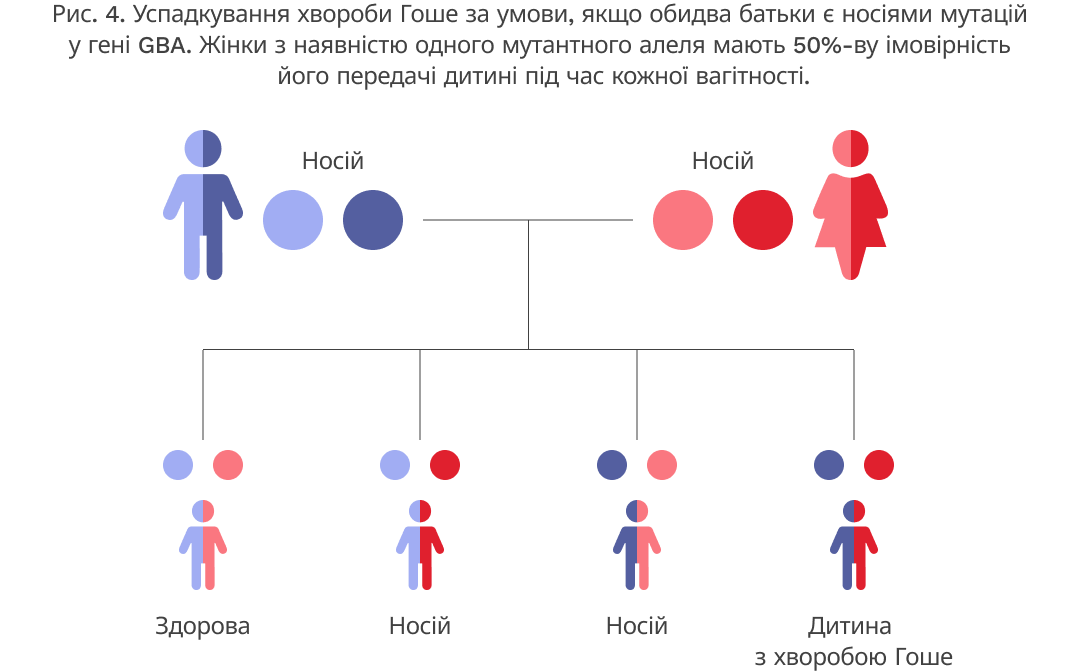
Хвороба Гоше є аутосомно-рецесивним захворюванням, це означає, що для його прояву обидві копії гена мають містити мутації. Батьки пацієнта з хворобою Гоше мають по одній копії мутованого гена GBA (є носіями), але вони, як правило, не проявляють ознак та симптомів хвороби. У кожного носія захворювання існує 50%-ва імовірність передачі мутантного варіанта гена при кожній вагітності. Якщо обидва батьки є носіями захворювання, то існує 25%-ва імовірність того, що дитина буде здоровою, і 25%-ва імовірність того, що дитина матиме хворобу Гоше (Рис. 4).5-8
При хворобі Гоше було виявлено понад 300 мутантних алелей у гені GBA.9 Клінічні прояви хвороби Гоше, як правило, неможливо точно передбачити на підставі виключно генетичного аналізу. Для більшості мутацій кореляція між генотипом і фенотипом є слабкою, однак для чотирьох мутантних алелей існують більш чіткі кореляції між генотипом і фенотипом, в тому числі наступні:
- Мутації N370S і 84GG переважно є основною причиною порівняно високої частоти виникнення захворювання серед євреїв-ашкеназі (приблизно до 1 на 850 осіб).3,4
- Мутація N370S пов'язана з хворобою 1 типу і виключає виникнення нейропатичного ураження.3
- Мутація L444P, виявлена у пацієнтів з провінції Норрботтен в Швеції, є прогностичним фактором виникнення нейропатичного захворювання 3 типу.4
- Мутація D409H тісно пов'язана із захворюванням типу 3с, при якому страждає функція серця.4
Відсутність у одного з батьків мутантного алеля гену GBA не є гарантією того, що дитина не матиме хворобу Гоше, адже у науковій літературі описано декілька випадків de novo мутацій у статевих клітинах.10,11
Література:
1. Mistry PK, Cappellini M, Lukina E, et al. Consensus Conference: a reappraisal of Gaucher disease - diagnosis and disease management algorithms. Am J Hematol 2011;86(1):110-115
2. Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105(1-2):151-156. doi:10.1007/s004399900075
3. Davies EH, Surtees R, DeVile C, Schoon I, Vellodi A. A severity scoring tool to assess the neurological features of neuronopathic Gaucher disease. J Inherit Metab Dis. 2007 Oct;30(5):768-82.
4. Dahl N, Lagerstrom M, Erikson A, Pettersson U. Gaucher disease type III (Norbottnian type) is caused by a single mutation in exon 10 of the acid-P glucosidase gene. Am J Hum Genet 1990;47(2):275-8.
5. Beutler E, Grabowski GA. Glucosylceramide lipidosis–Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited diseases. 8th ed. New York: McGraw-Hill; 2001. pp. 3635–68.
6. Tayebi N, Stone DL, Sidransky E. Type 2 gaucher disease: an expanding phenotype. Mol Genet Metab. 1999;68:209–19.
7. Gaucher disease-Genetics Home Reference. Reviewed September 2014. Available at: https://ghr.nlm.nih.gov/condition/gaucher-disease Last accessed July 02, 2020.
8. Gaucher disease. Genetic and Rare Diseases Information Center. Available at: https://rarediseases.info.nih.gov/diseases/8233/gaucher-disease. Last accessed July 02, 2020.
9. Koprivica V, Stone DL, Park JK, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66(6):1777-1786. doi:10.1086/302925
10. Saranjam H, Chopra SS, Levy H, et al. A germline or de novo mutation in two families with Gaucher disease: implications for recessive disorders. Eur J Hum Genet. 2013;21(1):115-117. doi:10.1038/ejhg.2012.105
11. Hagege E, Grey RJ, Lopez G, Roshan Lal T, Sidransky E, Tayebi N. Type 2 Gaucher disease in an infant despite a normal maternal glucocerebrosidase gene. Am J Med Genet A. 2017;173(12):3211-3215. doi:10.1002/ajmg.a.38487


