
How to Diagnose MPS I & II Disease
Clinical trial summaries
ENT and mucopolysaccharidoses
BIANCHI PM, GAINI R, VITALE S. ITALIAN JOURNAL OF PEDIATRICS 2018;44(SUPPL 2):127. HTTPS://DOI.ORG/10.1186/S13052-018-0555-0
Early diagnosis and management of cardiac manifestations in mucopolysaccharidoses: a practical guide for paediatric and adult cardiologists
BOFFI L, RUSSO P, LIMONGELLI G. ITALIAN J PEDIATRICS 2018;44(SUPPL 2):122.
Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis1
CIMAZ R, COPPA GV, KONÉ-PAUT I, ET AL. PEDIATRIC RHEUMATOLOGY 2009, 7:18 DOI:10.1186/1546-0096-7-18

Understanding Mucopolysaccharidosis type 1 (MPS I)
MPS I is a rare autosomal recessive disease caused by a deficiency of a-L-iduronidase, an enzyme required for the degradation of the glycosaminoglycans dermatan and heparan sulfate2
Due to the chronic and progressive accumulation of glycosaminoglycans in the lysosomes of cells throughout the body, patients affected by this devastating disease experience multiorgan dysfunction leading to considerable morbidity in most patients, and early mortality in those most severely affected. 2

MPS I has an incidence of approximately 1:100 000 and is panethnic with an equal sex incidence.

Patient photo courtesy of Dr Guelbert
Hurler, Hurler-Scheie, and Scheie syndromes
The MPS I spectrum is divided into phenotypes according to disease severity, rate of progression, age at presentation, and degree of cognitive involvement2
Signs and Symptoms of MPS I
Early symptom recognition and diagnosis are essential to achieve the best long-term prognosis in patients with MPS I. It is of paramount importance that not only pediatricians, but other specialists as well, are familiar with the clinical manifestations and consider an MPS diagnosis, especially when symptoms are present in combination with each other.2
Greater understanding of the symptomatology of the disease can lead to earlier diagnosis and initiation of treatment, which may in turn lead to better patient outcomes. Each of the three MPS I phenotypes, Hurler, Hurler–Scheie, and Scheie, is associated with a characteristic constellation of symptoms and disease course.2
Common misdiagnoses based on joint symptoms:6,8
- Rheumatoid arthritis
- Juvenile Idiopathic rheumatoid arthritis
- Growing pains
- Arthrogryiposis
- Muscular dystrophy
- Connective tissue disease
- Fibromyalgia
- Polyneuropathy
- Autoimmune disease

Most common signs and symptoms

Signs and Symptoms2


Adapted from Beck et al. Genetics in Medicine, 2014.
Prevalence and age of onset of signs and symptoms in patients with MPS I by phenotype. (a) Hurler, (b) Hurler–Scheie, and (c) Scheie. Percentages of symptom frequency are shown on the left axis. Only symptoms reported during the natural history period in at least 25% of patients with the relevant phenotype are shown. Age data are median ages in years (right axis) for those patients with the date of symptom onset recorded. MPS I, mucopolysaccharidosis type I.

Hernias are the earliest presenting symptom. Therefore the presence of inguinal or umbilical hernias in young children should raise suspicion of MPS I. 2
Should you wish to test for MPS I, kindly refer them to your local lab for a DBS (Dried Blood Spot) Test.
For any DBS sample collections, kit requests or diagnostic related queries, kindly contact:
Email: Ansie.Mienie@nwu.ac.za / pliem@nwu.ac.za
Contact Numbers: 018 299 2312 / 018 285 2544
WhatsApp Number: 082 393 8505
- Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. 2011 Dec 1;50(suppl_5):v4-12.
- Beck M, Arn P, Giugliani R, Muenzer J, Okuyama T, Taylor J, Fallet S. The natural history of MPS I: global perspectives from the MPS I Registry. Genetics in Medicine. 2014 Oct 1;16(10):759-65.
- Vijay S, Ed Wraith J. Clinical presentation and follow‐up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Pædiatrica. 2005 Jul;94(7):872-7.
- Barone R at al. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Italian journal of pediatrics. 2018 Nov;44(2):107-15.
- Giugliani R et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genetics and molecular biology. 2010;33(4):589-604.
- Cimaz R, Vijay S, Haase C, Coppa GV, Bruni S, Wraith E, Guffon N. Attenuated type I mucopolysaccharidosis in the differential diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Clinical and experimental rheumatology. 2006 Mar 1;24(2):196-202..
- Kubaski F, de Oliveira Poswar F, Michelin-Tirelli K, Matte UD, Horovitz DD, Barth AL, Baldo G, Vairo F, Giugliani R. Mucopolysaccharidosis type I. Diagnostics. 2020 Mar 16;10(3):161.
- Cimaz R, Coppa GV, Koné-Paut I, Link B, Pastores GM, Elorduy MR, Spencer C, Thorne C, Wulffraat N, Manger B. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatric Rheumatology. 2009 Dec 1;7(1):18.

Understanding Mucopolysaccharidosis type 1 (MPS I)
Mucopolysaccharidosis type II (MPS II) is a rare, life-limiting, X-linked recessive disease characterised by deficiency of the lysosomal enzyme iduronate-2-sulfatase.1
Like other mucopolysaccharidoses, the enzyme deficiency in MPS II results in the lysosomal accumulation of glycosaminoglycans (GAGs).1
MPS II occurs in all ethnic groups with an estimated incidence of 1: 140,000 to 333,000 births.2 The disease affects males almost exclusively, although a few symptomatic females have been identified.1
MPS II is a progressive disorder that has traditionally been categorised into a severe form and a mild/attenuated form based on the age at onset of signs and symptoms, the presence or absence of neurological involvement, and length of survival.1
The progressive accumulation of the GAG within tissues and organs is responsible for the clinical disease seen in MPS II.3

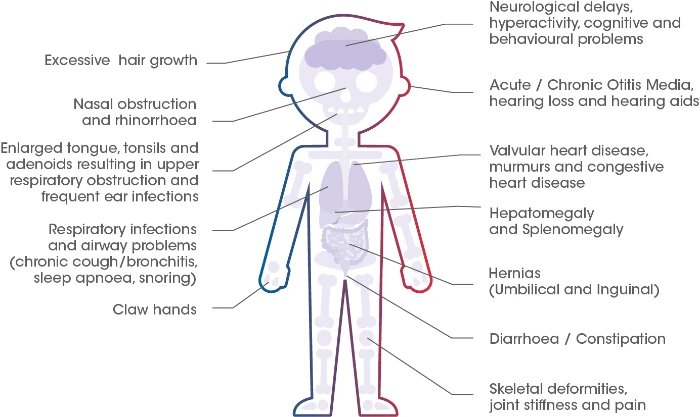
Common Signs and Symptoms of MPS II2,3

Although the specific combination of signs and symptoms may vary considerably between individuals, the presence of any of the features listed may be suggestive of MPS II. 1
The evolution of signs and symptoms is often a better indicator of a diagnosis of MPS II than a static snapshot of the presence or absence of certain manifestations. Therefore, it is important to monitor changes in signs and symptoms over time.1
Should you wish to test for MPS II, kindly refer them to your local lab for a DBS (Dried Blood Spot) Test.
For any DBS sample collections, kit requests or diagnostic related queries, kindly contact:
Email: Ansie.Mienie@nwu.ac.za / pliem@nwu.ac.za
Contact Numbers: 018 299 2312 / 018 285 2544
WhatsApp Number: 082 393 8505
- Scarpa M, Almássy Z, Beck M, Bodamer O, Bruce IA, De Meirleir L, Guffon N, Guillén-Navarro E, Hensman P, Jones S, Kamin W. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet journal of rare diseases. 2011 Dec;6:1-8.
- Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J, HOS Investigators. Initial report from the Hunter outcome survey. Genetics in Medicine. 2008 Jul 1;10(7):508-16.
- Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Molecular genetics and metabolism. 2007 Mar 1;90(3):329-37.