Mucopolysaccharidose I (MPSI)

MPS I is een zeldzame autosomale recessieve lysosomale stapelingsziekte veroorzaakt door een tekort aan het lysosomale enzym alfa-L-iduronidase (IDUA) dat betrokken is bij de afbraak van glycosaminoglycanen (GAGs): heparan- en dermatansulfaat. Lysosomale accumulatie van GAGs resulteert in disfunctie van cellen, weefsels en organen, zoals het hoornvlies, kraakbeen, bot, CZS en het bindweefsel van de huid, fascie, hartkleppen en bloedvaten.
MPS I is traditioneel onderverdeeld in drie fenotypen:
- het ernstige Hurler (MPS I-H)-fenotype,
- het intermediaire Hurler-Scheie (MPS I-H/S)-fenotype,
- en het mildere Scheie (MPS I/S)-fenotype.
Feitelijk vertoont MPS I echter een continu spectrum van fenotypische ernst. De geschatte incidentie van MPS I is 1/100.0001-4

De diagnose
Het onderzoek van de alfa-L-iduronidase (IDUA) enzymatische activiteit kan worden uitgevoerd op een Dried Blood Spot (DBS) of op volbloedstaal. Een verlaagde enzymatische activiteit moet worden gevolgd door genotypering om de diagnose van MPS I te bevestigen. Bevestiging van de verlaagde IDUA enzymatische activiteit op leukocyten is noodzakelijk voor de terugbetaling van de behandeling in België.
Indien u het vermoeden heeft van MPS, gelieve uw patient door te verwijzen naar één van de Belgische metabole centra (CEMA).

* Wanneer een MPS I of een andere MPS-stoornis wordt vermoed, kan een urinaire glycosaminoglycaan (uGAG)-analyse (zowel kwantitatief als kwalitatief) worden uitgevoerd. Een verhoogd uGAG-niveau en/of een abnormaal uGAG-patroon bevestigt de aanwezigheid van een MPS-aandoening en specifiek enzymonderzoek bepaalt het MPS-type.
Indien deze eerstelijnstesten niet resulteren in een diagnose en de patiënt symptomen (zoals groeiachterstand, skeletdysplasie of mentale retardatie) vertoont die geassocieerd zijn met verschillende genetische aandoendingen, is het mogelijk om meerdere genen tegelijk te analyseren met behulp van Next Generation Sequencing (NGS) technieken, zoals een genenpanel.
Gelieve een genetisch centrum te contacteren voor meer info.
De opsporingstest in de praktijk
Hieronder kan u praktische informatie terugvinden betreffende het uitvoeren van de opsporingstest via DBS of bloedstaal.
-(1).jpg/jcr:content/image%20(2)%20(1).jpg)
How to perform a DBS
De symptomen2-4
- Corneatroebeling
- Hepatosplenomegalie
- Hart(klep)afwijkingen
- Groeiachterstand
- Skeletafwijkingen
- Gewrichtscontracturen
- Carpaal tunnel syndroom, klauwhand
- Recidiverende navel- en/of liesbreuken
- Luchtwegobstructie
- Recidiverende keel-, neus- en oorinfecties
- Alleen bij Hurler/Hurler-Scheie: mogelijk grove gelaatstrekken, mogelijk mentale retardatie

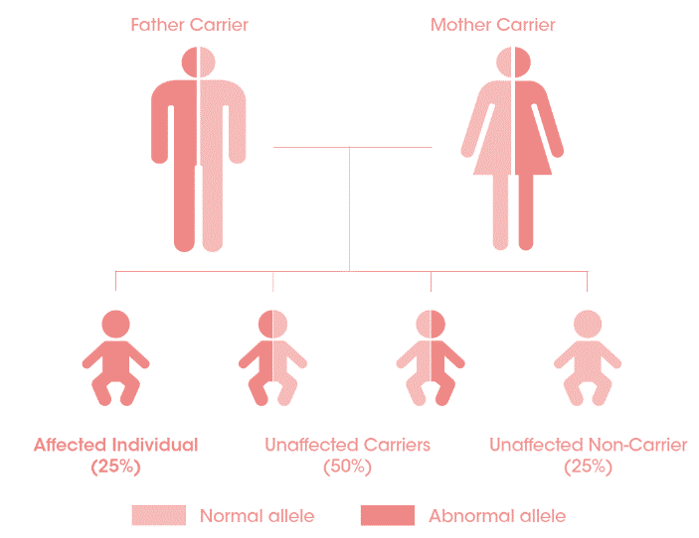
Broers en zussen testen
Test de patient, test de broers en zussen zo vroeg mogelijk:
indien een ouder drager is van de ziekte, heeft ieder kind 25% risico om getroffen te zijn door de ziekte5.
Product: Aldurazyme®
Aldurazyme® is geïndiceerd voor langdurige enzymsubstitutietherapie bij patiënten met een bevestigde diagnose van mucopolysaccharidose I (MPS I; α-L-iduronidasedeficiëntie) ter behandeling van de niet-neurologische manifestaties van de ziekte.
1. Neufeld EF, et al. The Online Metabolic and Molecular Bases of Inherited Disease New York, NY: McGraw-Hill; 2014 https://ommbid.mhmedical.com/content.aspx?sectionid=62642135&bookid=971&Resultclick=2. Accessed August 12, 2019
2. Beck, et al. Genet Med 2014;16(10):756–765
3. Cimaz R, et al. Pediatr Rheumatol Online J 2009;7:18
4. Guffon N, et al. Eur J Pediatr 2019;178(4):593-603
5. Clarke L. GeneReviews. February 11, 2016.
Bevestig uw status
Bevestig uw status
Sanofi biedt u exclusieve content over onze verschillende therapeutische gebieden op één portal
speciaal voor professionals in de gezondheidszorg, inclusief wetenschappelijk nieuws, een ljst met
evenementen en tools en diensten voor uw dagelijkse praktik.