Ziekte van gaucher

De ziekte van gaucher is een autosomale recessieve lysosomale stapelingsziekte die wordt veroorzaakt door een tekort aan of verminderde activiteit van zure β-glucosidase (GBA-1) die resulteert in de accumulatie van zijn substraat glucocerebroside (GL-1). De accumulatie van GL-1 in de lysosomen van macrofagen leidt tot progressieve hepatosplenomegalie, hematologische abnormaliteiten zoals bloedarmoede en trombocytopenie, botziekte en andere manifestaties.
De ziekte van gaucher is een pan-etnische ziekte met een totale incidentie van 1/40.000 -1/60.000, maar kan oplopen tot 1/850 in asjkenazisch joodse bevolkingsgroepen.1-5

De diagnose
Een definitieve diagnose van de ziekte van gaucher wordt bevestigd door het aantonen van een tekort aan zure β-glucosidaseactiviteit in leukocyten en/of door de aanwezigheid van 2 mutaties in het GBA-gen. Een andere LSD genaamd ASMD (Acid Sphingomyelin Maltase Deficiency) heeft meerdere klinische manifestaties gemeen met de ziekte van Ggaucher. Daarom wordt (sterk) aanbevolen om gelijktijdig enzymonderzoek voor zure β-glucosidase (GBA-1) en zure sfingomyelinase (ASM) uit te voeren.1-5
Indien u het vermoeden heeft van de ziekte van gaucher, gelieve uw patient door te verwijzen naar één van de Belgische metabole centra (CEMA).
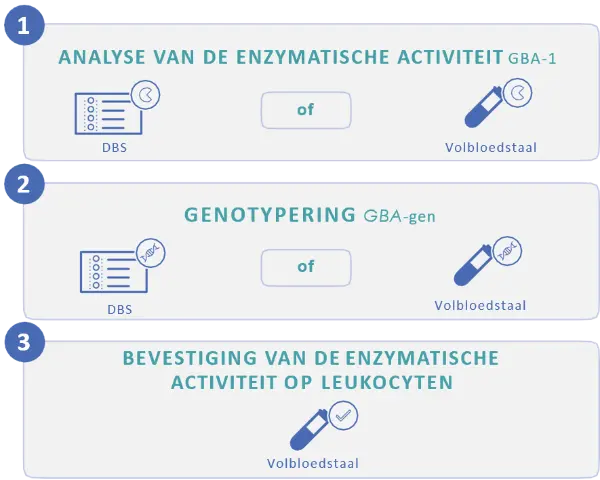
Indien deze eerstelijnstesten niet resulteren in een diagnose en de patiënt symptomen (zoals splenomegalie, trombocytopenie of groeiachterstand) vertoont, die geassocieerd zijn met verschillende genetische aandoendingen, is het mogelijk om meerdere genen tegelijk te analyseren met behulp van Next Generation Sequencing (NGS) technieken, zoals een genenpanel.
Gelieve een genetisch centrum te contacteren voor meer info.
De opsporingstest in de praktij
Hieronder kan u praktische informatie terugvinden betreffende het uitvoeren van de opsporingstest via DBS of bloedstaal.
-(1).jpg/jcr:content/image%20(2)%20(1).jpg)
How to perform a DBS
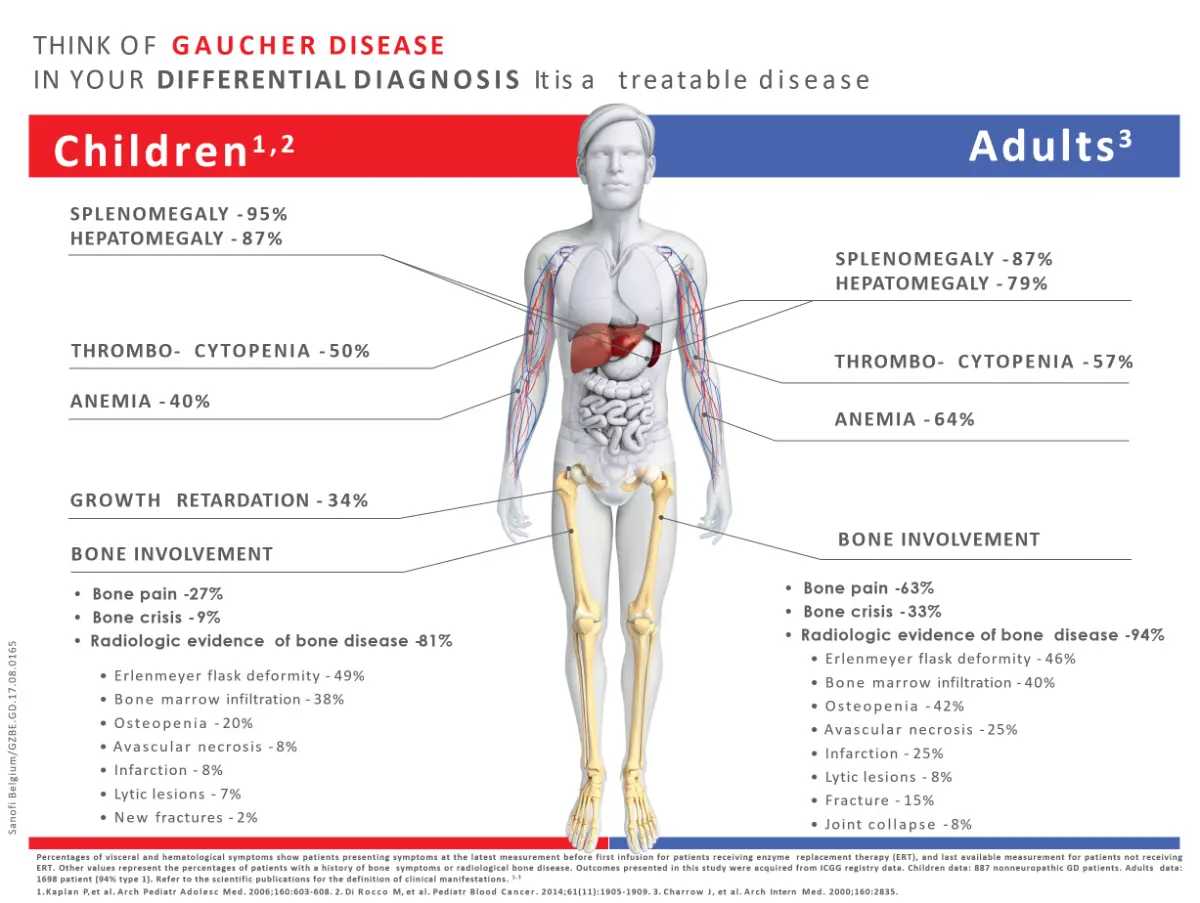
De symptomen
- Hepatosplenomegalie
- Botpijn, botcrisis
- Bloedarmoede, trombocytopenie
- Vermoeidheid
- Erlenmeyer flask deformity
- Osteopenie, pathologische fracturen
- Beenmerginfiltratie door Gaucher cellen
- Avascularie necrose
- Groeiachterstand, verlate puberteit1,3,5
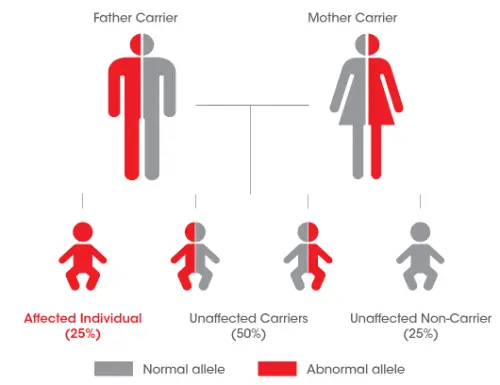
Broers en zussen testen
Test de patient, test de broers en zussen zo vroeg mogelijk: indien een ouder drager is van de ziekte, heeft ieder kind 25% risico om getroffen te zijn door de ziekte.6
Producten: Cerdelga® en Cerezyme®
Sanofi Genzyme is al meer dan 30 jaar betrokken bij de gaucher patiënten. Het allereerste substitutie enzym van humane placenta's werd gelanceerd in 1991: Ceredase®.
Verdere onderzoeksinspanningen leidden vervolgens tot de lancering van het recombinant enzym Cerezyme® in 1994. In 2015 werd de eerste orale behandeling gelanceerd voor volwassen Gaucher type 1 patiënten: een eerste lijn substraat inhibitor: Cerdelga®.

.svg)
Materialen
Alles
Voor HCP
Voor Patiënten
- Charrow J, et al. Arch Intern Med. 2000;160:2835
- Kaplan, Paige, et al. “Revised recommendations for the management of Gaucher disease in children.” European journal of pediatrics 172.4 (2013): 447-458
- Di Rocco, M. et al. Early diagnosis of Gaucher disease in pediatric patients: proposal for a diagnostic algorithm. Pediatr. Blood Cancer 61, 1905 -1909 (2014)
- Mistry, Pramod K., et al. `Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists -oncologists and an opportunity for early diagnosis and intervention.’ American journal of hematology 82.8 (2007): 697-701
- McGovern, Margaret M., et al. “Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency.” Genetics in Medicine (2017)
- Orphaschool. [En ligne] Orphanet. http://www.orpha.net/orphaschool/formations/transmission/ExternData/InfoTransmission-Dreamweaver/Transmission.pdf



Bevestig uw status
Bevestig uw status
Sanofi biedt u exclusieve content over onze verschillende therapeutische gebieden op één portal
speciaal voor professionals in de gezondheidszorg, inclusief wetenschappelijk nieuws, een ljst met
evenementen en tools en diensten voor uw dagelijkse praktik.