Maladie de Gaucher

Maladie de Gaucher
La maladie de Gaucher est une maladie de surcharge lysosomale autosomique récessive due à l’activité déficiente ou réduite de l’enzyme β-glucosidase acide (GBA-1) qui induit l’accumulation de son substrat, le glucocérébroside (GL-1). L’accumulation de GL-1 dans les lysosomes des macrophages entraîne une hépatosplénomégalie progressive, des anomalies hématologiques y compris une anémie et une thrombocytopénie, une maladie osseuse et d’autres manifestations.
La maladie de Gaucher est une maladie pan-ethnique dont l’incidence globale s’élève de 1/40 000 à 1/60 000 mais peut atteindre 1/850 dans la population juive ashkénaze.1-5

Le diagnostic
Le diagnostic définitif de la maladie de Gaucher est confirmé par une mesure de l’activité de la β-glucosidase acide dans les leucocytes et/ou par la présence de 2 mutations dans le gène GBA.
Une autre maladie de surcharge lysosomale appelée ASMD (Acid Sphingomyelin Maltase Deficiency) a plusieurs manifestations cliniques en commun avec la maladie de Gaucher. Il est donc (vivement) recommandé d’effectuer des tests enzymatiques simultanés pour la β-glucosidaseacide et la sphingomyélinase acide (ASM).1-5
En cas de suspicion de maladie de Gaucher, veuillez référer votre patient vers l’un des centres belges de référence métabolique (CEMA).

Si ces tests de première ligne ne permettent pas de poser un diagnostic et que le patient présente des symptômes (tels que splénomégalie, thrombocytopénie ou retard de croissance) associés à différentes affections génétiques, il est possible d’analyser plusieurs gènes simultanément au moyen de techniques Next Generation Sequencing (NGS), telles qu’un panel de gènes.
Veuillez contacter un centre génétique pour obtenir de plus amples informations.
Le test de dépistage en pratique
Retrouvez ci-dessous les informations pratiques pour réaliser le test de dépistage sur DBS ou sur sang complet.
-(1).jpg0/jcr:content/image%20(2)%20(1).jpg)
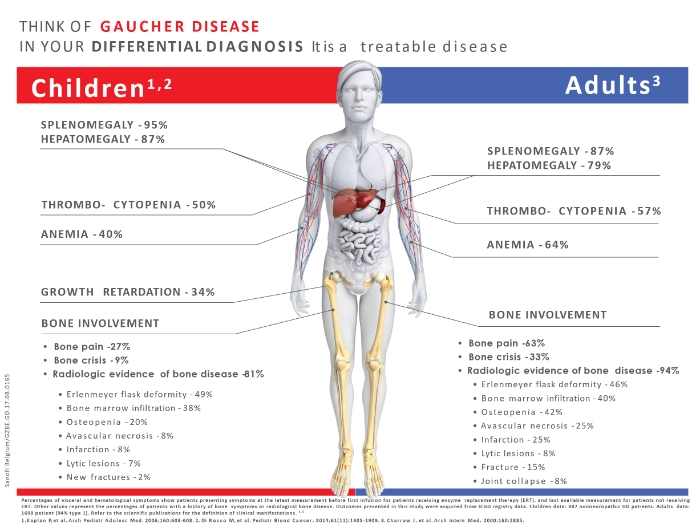
Les symptômes
- Hépatosplénomégalie
- Douleurs osseuses, crises osseuses
- Anémie, thrombocytopénie
- Fatigue
- Déformation de l’extrémité des os longs en flacon d’Erlenmeyer
- Ostéopénie, fractures pathologiques
- Infiltration médullaire par les cellules de Gaucher
- Nécrose avasculaire
- Retard de croissance, puberté retardée1,3,5
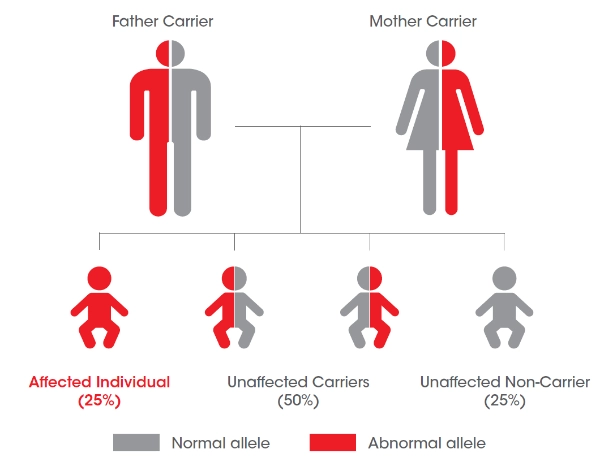
Le dépistage de la fratrie
Testez le patient, testez la fratrie et diagnostiquez les patients au plus tôt : pour un parent porteur de la maladie, chaque enfant présente 25% de risque d’être touché par la maladie.6
Produits: Cerdelga® et Cerezyme®
Sanofi Genzyme s’engage auprès de la communauté des patients atteints de la maladie de Gaucher depuis plus de 30 ans. La toute première enzyme de substitution issue de placentas humains a été lancée en 1991 : le Ceredase®.
Les efforts continus de recherche ont ensuite mené au lancement de l’enzyme recombinante Cerezyme® en 1994. Puis, en 2015, le premier traitement oral inhibiteur de substrat de première ligne pour les patients adultes atteints de la maladie de Gaucher de type 1 a vu le jour : le Cerdelga®.

.svg)
Matériels
𝗧𝗼𝘂𝘁
𝗣𝗼𝘂𝗿 𝗛𝗖𝗣
- Charrow J, et al. Arch Intern Med. 2000;160:2835
- Kaplan, Paige, et al. “Revised recommendations for the management of Gaucher disease in children.” European journal of pediatrics 172.4 (2013): 447-458
- Di Rocco, M. et al. Early diagnosis of Gaucher disease in pediatric patients: proposal for a diagnostic algorithm. Pediatr. Blood Cancer 61, 1905 -1909 (2014)
- Mistry, Pramod K., et al. `Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists -oncologists and an opportunity for early diagnosis and intervention.’ American journal of hematology 82.8 (2007): 697-701
- McGovern, Margaret M., et al. “Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency.” Genetics in Medicine (2017)
- Orphaschool. [En ligne] Orphanet. http://www.orpha.net/orphaschool/formations/transmission/ExternData/InfoTransmission-Dreamweaver/Transmission.pdf



Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.