Maladie de Pompe
.png/jcr:content/hero%20image%20muscle%20road%20-%201400x550%20(1).png)
La maladie de Pompe
Maladie de Pompe est une maladie de surcharge lysosomale autosomique récessive due à des mutations du gène codant pour l’a-glucosidase acide (GAA) ou maltase acide. L’accumulation de son substrat, le glycogène, est associée à un large spectre de symptômes cliniques et à des degrés variables de progression, d’apparition des symptômes, d’atteinte organique et de sévérité. L'accumulation de glycogène se produit dans divers tissus, mais le tissu musculaire squelettique, le cœur et les muscles lisses sont les plus affectés. Les patients les plus gravement atteints ont une activité GAA réduite ou absente et décèdent généralement au cours de la première année de vie. Les patients moins gravement atteints peuvent ne pas être diagnostiqués avant d’avoir atteint l’âge adulte. L’incidence de la maladie de Pompe est estimée à 1/40.000 1-5
.png)
Le diagnostic
L’activité enzymatique peut être déterminée par l’analyse sur Dried Blood Spot test (DBS). Un déficit en a-glucosidase acide dans une culture de fibroblastes cutanés et une analyse génétique ADN mettant en évidence deux mutations pour le gène GAA sont nécessaires pour obtenir le remboursement du traitement en Belgique.
Une accumulation lysosomale de glycogène doit être détectée dans une biopsie musculaire si une seule mutation GAA pathologique du gène est observée.1-5
En cas de suspicion de maladie de Pompe, veuillez référer votre patient vers l’un des centres belges neuromusculaire de référence (NMRC) ou un des centres de référence métabolique (CEMA).
.png)
*Le statut de CRIM (Cross-Reactive Immunological Material) se réfère à la présence ou à l’absence de protéine GAA endogène avec ou sans activité enzymatique. Les patients CRIM-négatifs ne produisent pas de protéine GAA fonctionnelle propre et ont été rapportés comme ayant un pronostic moins bon que les patients CRIM-positifs.
Retrouvez ci-dessous les informations pratiques pour réaliser l’analyse CRIM.
Si ces tests de première ligne ne permettent pas de poser un diagnostic et que le patient présente des symptômes (tels que splénomégalie, thrombocytopénie ou retard de croissance) associés à différentes affections génétiques, il est possible d’analyser plusieurs gènes simultanément au moyen de techniques Next Generation Sequencing (NGS), telles qu’un panel de gènes.
Veuillez contacter un centre génétique pour obtenir de plus amples informations.
Le test de dépistage en pratique
Retrouvez ci-dessous les informations pratiques pour réaliser le test de dépistage sur DBS ou sur sang complet.
-(2).jpg/jcr:content/image%20(2)%20(2).jpg)
How to perform a DBS
Les symptômes
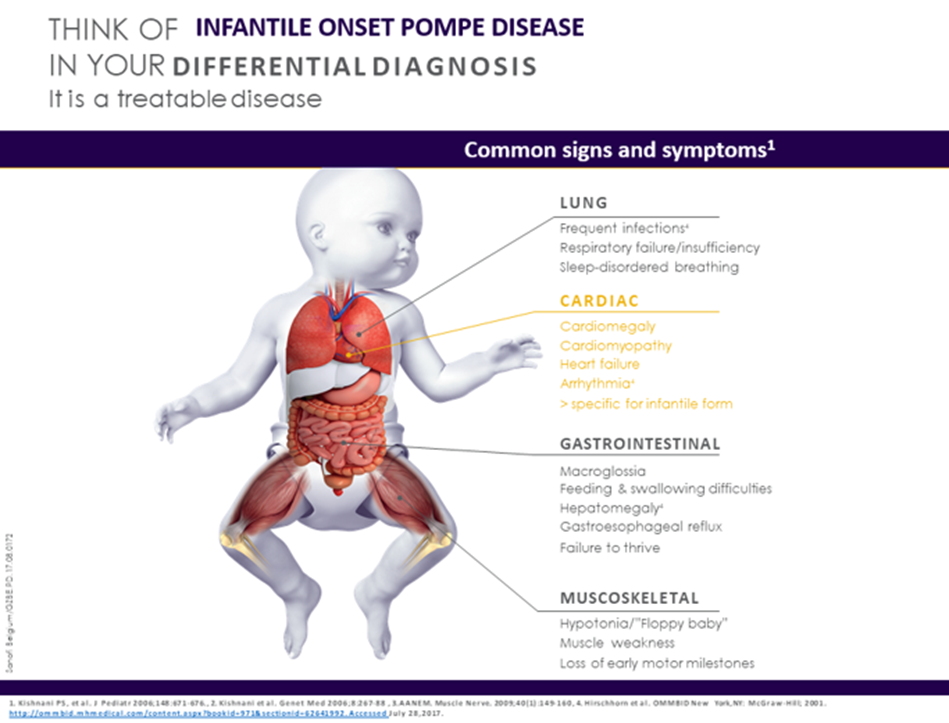
Maladie de Pompe infantile 4
- Défaillance/insuffisance respiratoire, infections respiratoires fréquentes
- Troubles respiratoires du sommeil
- Cardiomégalie, cardiomyopathie, insuffisance cardiaque, arythmie
- Problèmes d’alimentation et de déglutition, macroglossie
- Retard staturo-pondéral, retard du développement moteur
- Hypotonie/bébé « mou », faiblesse musculaire
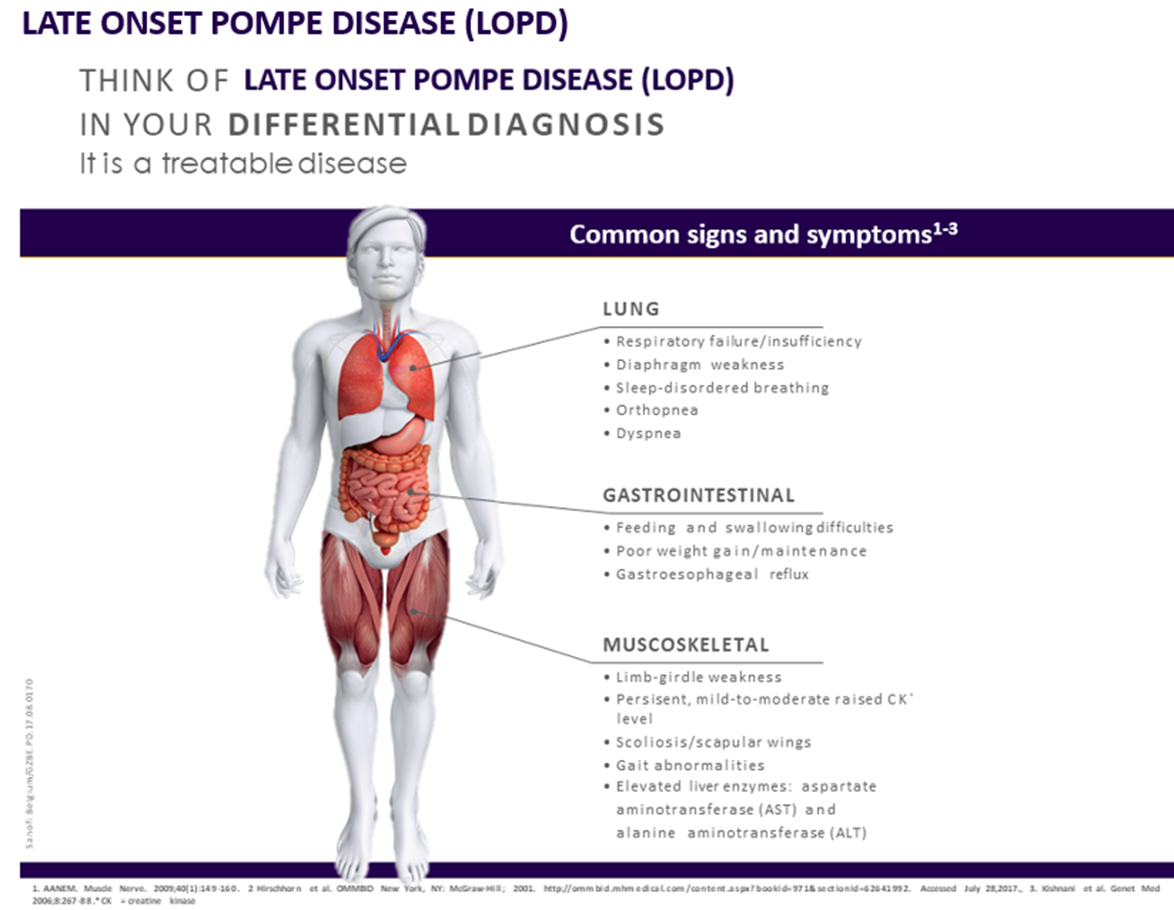
Maladie de Pompe tardive 3
- Défaillance/insuffisance respiratoire, faiblesse du diaphragme, troubles respiratoires du sommeil, orthopnée, dyspnée
- Problèmes d’alimentation et de déglutition, faible augmentation/maintien du poids, reflux gastro-œsophagien
- Faiblesse au niveau des ceintures
- Scoliose/scapula alata
- Démarche anormale, problèmes pour se lever (signe de Gowers)
- Enzymes hépatiques élevées : aspartate aminotransférase (ASAT) et alanine aminotransférase (ALAT)
- Taux de créatine kinase systématiquement légèrement à modérément élevé
Le dépistage de la fratrie
Testez le patient, testez la fratrie et diagnostiquez les patients au plus tôt : pour un parent porteur de la maladie, chaque enfant présente 25% de risque d’être touché par la maladie.9
.png)
Matériels
𝗧𝗼𝘂𝘁
𝗣𝗼𝘂𝗿 𝗛𝗖𝗣
𝗣𝗼𝘂𝗿 𝗹𝗲𝘀 𝗽𝗮𝘁𝗶𝗲𝗻𝘁𝘀
- Hirschhorn et al. OMMBID New York, NY: McGraw-Hill; 2001 http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62641992. Accessed August 12, 2019
- Kishnani PS, et al. Genet Med 2006;8:267-88
- Kishnani PS, et al. Mol Genet Metab. Jan 2010;99(1):26-33
- Kishnani PS, et al. J Pediatr 2006;148:671-676
- AANEM. Muscle Nerve. 2009;40(1):149-1602
- Johnson K, et al. Orphanet J Rare Dis 2017;12(1):173
- Lévesque S, et al. Orphanet J Rare Dis 2016; 11:8
- Nallamilli BRR, et al. Ann Clin Transl Neurol 2018; 1;5(12):1574-1587
- Toscano et al., Acta Myol. 2013; 32(2): 78-81

Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.