La mucopolysaccharidose de type I (MPS I)

La MPS I est une maladie de surcharge lysosomale autosomique récessive due à un déficit de l’enzyme lysosomale alpha-L-iduronidase (IDUA) qui intervient dans la dégradation des glycosaminoglycanes (GAG) : héparane sulfate et dermatane sulfate. L’accumulation lysosomale de GAG induit des dysfonctions cellulaires, tissulaires et organiques tels que la cornée, le cartilage, les os, le SNC et le tissu conjonctif de la peau, les fascias, les valves cardiaques et les vaisseaux sanguins.
La MPS I est traditionnellement subdivisée en trois phénotypes :
- le phénotype Hurler (MPS I-H) sévère,
- le phénotype Hurler-Scheie (MPS I-H/S) intermédiaire,
- et le phénotype Scheie (MPS I/S) atténué.
En réalité, MPS I présente un spectre continu de gravité phénotypique. L’incidence de la maladie de MPS I est estimée à 1/100.0001-4.

Le diagnostic
L’examen des niveaux d’activité enzymatique de l’alpha-L-iduronidase (IDUA) peut être effectué par Dried Blood Spot (DBS) test ou sur sang complet. La mise en évidence d’un déficit marqué en IDUA doit être suivi d’un génotypage pour confirmer la MPS I. Des tests pour confirmer l’activité enzymatique IDUA sur leucocytes sont ensuite nécessaires pour obtenir le remboursement du traitement en Belgique.
En cas de suspicion de MPS, veuillez référer votre patient vers l’un des centres belges de référence métabolique (CEMA).
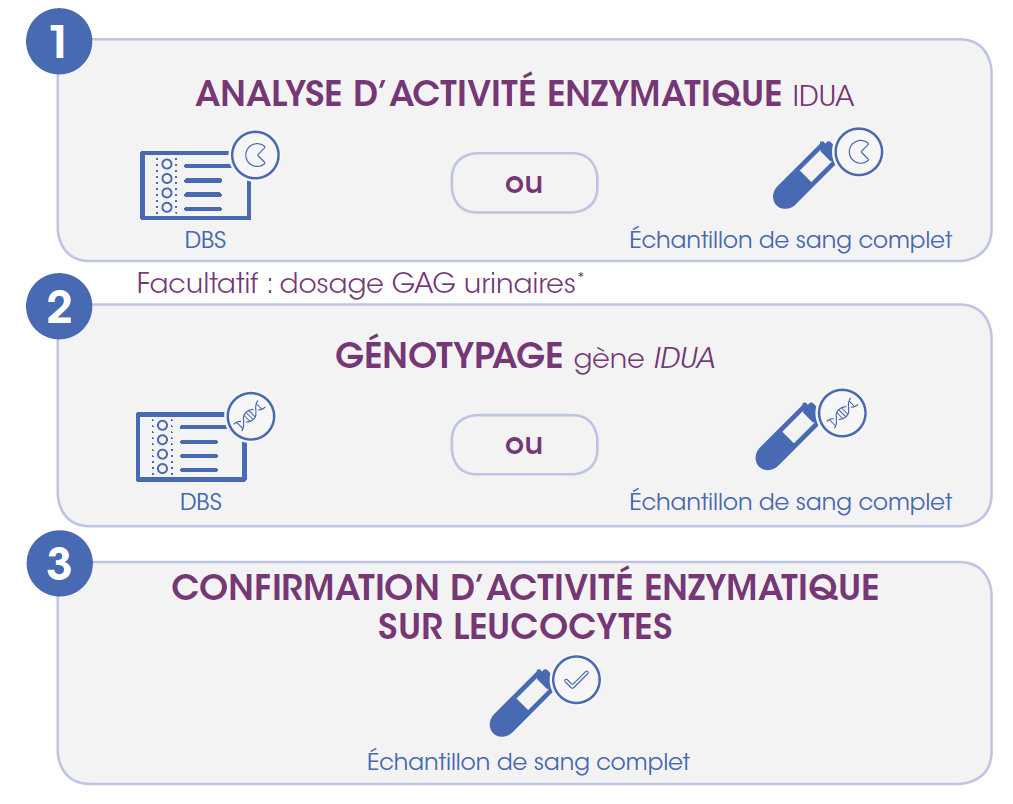
* En cas de suspicion d’une MPS I ou d’une autre MPS, un dosage des glycosaminoglycanes urinaires (uGAG) (tant quantitatif que qualitatif) peut être effectué. Un taux d’uGAG élevé et/ou un schéma uGAG anormal confirme la présence d’un trouble MPS, et des tests enzymatiques spécifiques détermineront le type de MPS.
Si ces tests de première ligne ne permettent pas de poser un diagnostic et que le patient présente des symptômes (tels que retard de croissance, dysplasie squelettique ou retard mental) associés à différentes affections génétiques, il est possible d’analyser plusieurs gènes simultanément au moyen de techniques Next Generation Sequencing (NGS), telles qu’un panel de gènes.
uillez contacter un centre génétique pour obtenir de plus amples informations.
Le test de dépistage en pratique
Retrouvez ci-dessous les informations pratiques pour réaliser le test de dépistage sur DBS ou sur sang complet.
-(1).jpg1/jcr:content/image%20(2)%20(1).jpg)
Les symptômes2-4
- Opacification de la cornée
- Hépatosplénomégalie
- Anomalies (valvulaires) cardiaques
- Retard de croissance
- Anomalies squelettiques
- Contractures articulaires
- Syndrome du canal carpien, main en griffe
- Hernie ombilicale et/ou inguinale récidivante
- Obstruction des voies aériennes
- Infections ORL récidivantes
- Uniquement dans le syndrome Hurler/Hurler-Scheie : éventuellement traits du visage grossiers et retard mental

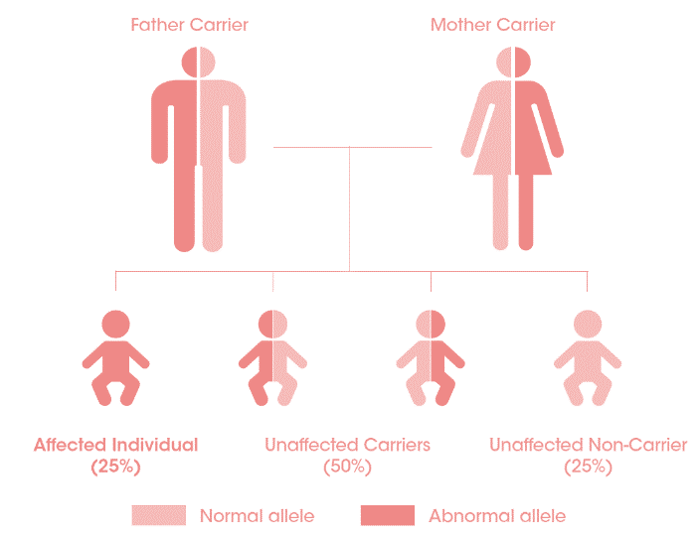
Le dépistage de la fratrie
Testez le patient, testez la fratrie et diagnostiquez les patients au plus tôt :
pour un parent porteur de la maladie, chaque enfant présente 25% de risque d’être touché par la maladie5.
Produit: Aldurazyme®
Aldurazyme® est indiqué en tant que traitement enzymatique substitutif à long terme chez les patients présentant un diagnostic confirmé de mucopolysaccharidose de type I (MPS I ; déficit d’α-L-iduronidase), afin de traiter les manifestations non neurologiques de la maladie.
- Neufeld EF, et al. The Online Metabolic and Molecular Bases of Inherited Disease New York, NY: McGraw-Hill; 2014 https://ommbid.mhmedical.com/content.aspx?sectionid=62642135&bookid=971&Resultclick=2. Accessed August 12, 2019
- Beck, et al. Genet Med 2014;16(10):756–765
- Cimaz R, et al. Pediatr Rheumatol Online J 2009;7:18
- Guffon N, et al. Eur J Pediatr 2019;178(4):593-603
- Clarke L. GeneReviews. February 11, 2016
Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.