
Seltene Krankheiten – Lysosomale Speicherkrankheiten
Sie sind Arzt/Ärztin, Medizinisches Fachpersonal
Wissenschaftliche Daten von medizinischem Interesse, die für Ärzte und andere Angehörige der Gesundheitsberufe bestimmt sind, sind auf dieser Website nach Anmeldung (SwissRX, DocCheck, Registrierung) verfügbar.
Sie sind Patient/in, Angehörige/r
Weitere Informationen zu lysosomalen Speicherkrankheiten erhalten Sie unter lysomed.ch, die Schweizer Informationsseite über seltene Krankheiten
Was sind seltene Krankheiten?
Eine Krankheit gilt als selten, wenn sie nicht mehr als 5 von 10’000 Personen betrifft. Obwohl die einzelnen Krankheitsbilder selten sind, ist die Gruppe mit 6’000 – 8’000 beschriebenen Krankheiten sehr gross.
Lysosomale Speicherkrankheiten
Beim Geschäftsbereich von Sanofi, der sich den seltenen Krankheiten widmet, konzentrieren wir uns auf die lysosomalen Speicherkrankheiten. Mittlerweile umfasst diese Gruppe mehr als 60 verschiedene Erkrankungen.
Sie gehen alle auf eine genetische Ursache zurück, die zu einem Mangel oder einer Dysfunktion bestimmter Enzyme führt. Ist das Enzym nicht (genügend) vorhanden, oder ist dessen Aktivität reduziert, können Zellabfälle nicht mehr hinreichend abgebaut werden. Dies führt zunächst zur Akkumulation der abzubauenden Substanzen in den Lysosomen (Zellorganellen) und langfristig zu Organschäden. Damit können unterschiedlichste Symptome ausgelöst werden.

Wer ist von lysosomalen Speicherkrankheiten betroffen?
Lysosomale Speicherkrankheiten sind erblich und können von den Eltern an ihre Kinder weitergegeben werden. Jeder Mensch bekommt von jedem Gen je eine Kopie von seiner Mutter und seinem Vater. Sind beide Gene fehlerfrei, wird im Körper ein funktionsfähiges Enzym hergestellt. Ist eines der Gene beschädigt (mutiert), kommt es darauf an, ob die Mutation dominant oder rezessiv ist und ob es sich auf einem Geschlechts-chromosom oder Autosom befindet. Durch all diese Faktoren wird das Erkrankungsrisiko der Nachkommen bestimmt.

Symptome, Krankheitsverlauf und Versorgung bei lysosomalen Speicherkrankheiten
Die Symptome der verschiedenen lysosomalen Speicherkrankheiten sind so unterschiedlich wie auch die Organe, die davon betroffen sind. Darum lösen manche der Krankheiten relativ wenige und eher leichte Symptome aus, während andere zu schwerwiegenden Problemen führen. Auch der Zeitpunkt des Auftretens der ersten Symptome variiert stark, sodass einige Patienten bereits im frühen Kindesalter betroffen sind, während andere erst in den späten Lebensjahren unter der Krankheit leiden.
Erfahren Sie mehr über die Symptomatik:
Dies führt leider sehr häufig dazu, dass lysosomale Speicherkrankheiten oft erst spät erkannt und diagnostiziert werden. Bei manchen Betroffenen dauert es Jahre oder gar Jahrzehnte, bis sie bei einem Facharzt für lysosomale Speicherkrankheiten landen.
Um das defiziente Enzym dem Körper wieder zuzuführen, gibt es verschiedene Therapiemöglichkeiten. Sanofi zeichnete sich als Pionier aus, als sie 1984 erstmals eine Enzymersatztherapie entwickelte. Informationen zu den verschiedenen Therapien finden Sie unter: www.lysomed.ch/de/lysosomale-speicherkrankheiten/behandlung

Tests für die Diagnose seltener Erkrankungen
Wenn aufgrund der Symptome der Verdacht besteht, dass es sich um eine lysosomale Speicherkrankheit handeln könnte, sollten Betroffene ihren Arzt informieren. Dieser wird zunächst häufigere Ursachen für die Symptome ausschliessen. Wenn sich weiterhin keine klärende Diagnose finden lässt, ist es höchste Zeit, auch an eine seltene Krankheit zu denken.

Ein einfacher Test kann Klarheit bringen
Für den Trockenbluttest wird Ihnen in der Arztpraxis Blut abgenommen und einige Tropfen auf eine spezielle Filterkarte aufgetragen. Wenn die Karte getrocknet ist, kann sie in einem speziellen Couvert per Post an das Labor geschickt werden. Im Labor wird dann das Blut wieder aus der Karte gelöst und die Aktivität des betroffenen Enzyms gemessen. Liegt sie unter dem Normwert, erhärtet sich die Verdachtsdiagnose.
Meist wird mit einem Gentest aus der gleichen Blutprobe die Mutation gesucht, um die Diagnose zu bestätigen.

MAT-CH-2201968-3.0 - 11/2025
fr-4
.jpg-400w.jpg-400w.png/jcr:content/diabete-photo.2022-01-31-19-33-00%20(1).jpg%20400w.jpg%20400w.png)
Evolution & prise en charge
Complications
Les risques et les complications possibles sont nombreux lorsque le diabète gestationnel n’est pas bien contrôlé.
Risques pour l’enfant
- Macrosomie : poids à la naissance supérieur à 4kg
- Hypoglycémie à la naissance
- Risque de blocage lors de la sortie des épaules pendant l’accouchement, ce qui peut engager le pronostic vital de l’enfant
- Risque plus élevé d’obésité, de diabète, d’hypertension et de maladie rénale à l’âge adulte
Risques pour la mère
- Accouchement prématuré du fait d’un surplus de liquide amniotique
- Accouchement par césarienne (à cause, entre autres, du poids du bébé)
- Hypertension de grossesse ou pré-éclampsie (tension artérielle élevée et enflure)
- Risque de développer un diabète de type 2 après la grossesse
Prise en charge
Généralement, une alimentation saine ainsi qu’une bonne hygiène de vie (gestion du stress, sommeil adéquat et activité physique) sont suffisantes pour contrôler le diabète de la femme enceinte.
Il peut arriver dans certains cas qu'un traitement à base d'insuline soit parfois nécessaire.
diabete-diabete-type2-3
Evolution & prise en charge
Évolution et complications
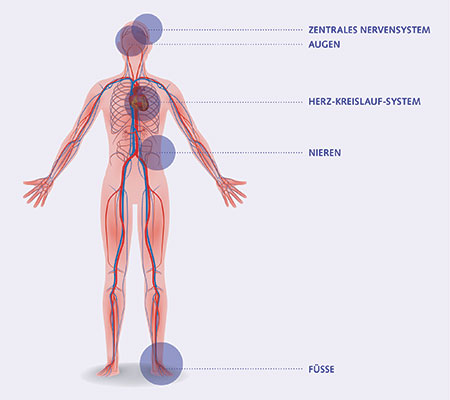
Lorsqu’ils ne sont pas correctement gérés, tous les types de diabète peuvent conduire à des complications en différents endroits de l’organisme. Celles-ci peuvent se manifester par des symptômes mais sont souvent silencieuses.
Des taux de glycémie en permanence élevés provoquent des dommages sur plusieurs organes et parties du corps, principalement :
• les reins (insuffisance rénale, néphropathie)
• les yeux (rétinopathie)
• le système nerveux (neuropathie)
• le cœur (infarctus)
• les vaisseaux sanguins (hypertension, artériosclérose, accident vasculaire cérébral [AVC], etc.)
• les pieds (pied diabétique) et les mains
Pendant la grossesse, un diabète mal géré augmente le risque de complications pour la mère et le fœtus.
Avec un contrôle régulier de la glycémie et un traitement individuel adéquat, il est possible de réduire le risque de survenue de ces complications.
Prise en charge
La base de tout traitement du diabète de type 2 est :
• une alimentation saine et équilibrée
• une activité physique
• une perte de poids
Si ces mesures n'apportent aucune amélioration de votre glycémie, le traitement peut être renforcé par des médicaments.
Traitement antidiabétique par voie orale (comprimés)
Un traitement antidiabétique par voie orale (comprimés) est recommandé si le pancréas peut encore produire suffisamment d'insuline. Ce traitement, en stimulant la sécrétion d'insuline dans les cellules bêta du pancréas ou la sensibilité des cellules à l'insuline, va favoriser l'entrée du glucose dans les cellules.
En Suisse, il existe plusieurs classes d’antidiabétiques oraux reposant sur des mécanismes d’action différents, à utiliser seules ou associées entre elles :
• Les biguanides comme la metformine,
• Les sulfonylurées,
• Les thiazolidinediones,
• Les inhibiteurs de DPP-4,
• Les inhibiteurs de la SGLT2
Traitement antidiabétique par injection
Les analogues du GLP-1
Les médicaments à base de GLP-1, appelés « analogues ou agonistes du récepteur au GLP-1 » (GLP-1 RA), peuvent contribuer à améliorer la glycémie dans le cadre du traitement du diabète de type 2.
Ces médicaments imitent ou renforcent la fonction naturelle de l’hormone intestinale GLP-1 présente dans le corps. Tout comme l'hormone naturelle du corps, les GLP-1 RA peuvent également stimuler la sécrétion d’insuline par le pancréas et ainsi réduire la glycémie. Avec un GLP-1 RA, la sécrétion d’insuline est moins stimulée lorsque la glycémie est basse et est plus stimulée lorsque la glycémie est élevée, ce qui est un avantage de ce mécanisme d’action. Par conséquent, le risque d’hypoglycémie avec un traitement à base de GLP-1 est faible. Tout comme l’hormone intestinale naturelle, ce type de médicament ralentit le transit gastro-intestinal et peut ainsi réduire l’appétit. La possibilité de perdre du poids est donc un autre avantage des GLP-1 RA.
Comme l’insuline, les GLP-1 RA sont des médicaments qui sont injectés sous la peau. L’injection est réalisée une fois par jour ou une fois par semaine selon le médicament.
Il est également à noter que les GLP-1 RA ont différents effets sur le profil glycémique. En effet, certains agissent principalement sur la glycémie à jeun, tandis que d’autres agissent sur la glycémie postprandiale (après un repas).
Insulines
Lorsque votre pancréas produit trop peu ou plus du tout d'insuline, ceci malgré une alimentation adaptée et/ou la prise de comprimés antidiabétiques ou d’un GLP-1 RA, la glycémie reste en permanence trop élevée. Ceci peut provoquer des troubles désagréables, mais également des complications dangereuses. Il faut donc faire baisser le taux de sucre dans le sang en prenant de l'insuline. L'insuline est une hormone qui peut uniquement être injectée sous la peau.
Il existe une large gamme de préparations à base d'insuline afin de permettre un traitement individuel et adapté à votre diabète.
L'insuline est répartie en 3 groupes principaux :
• L'insuline rapide agit très rapidement et uniquement pendant une courte période. Agissant principalement sur les glycémies postprandiales, elle est prise en association avec les repas.
• L'insuline basale agit au bout d'un certain temps seulement. Elle permet de couvrir les besoins fondamentaux du corps en insuline, qu’on appelle le taux basal et permet ainsi de réduire principalement la glycémie à jeun.
• Les insulines mixtes sont des mélanges fixes d'insulines rapides et d'insulines à effet retard.
Les différences prépondérantes entre les insulines sont la rapidité du passage de l'insuline dans le sang et la quantité d'insuline administrée. Lisez toujours la notice jointe à l'insuline.
Les combinaisons fixes d’insuline basale et de GLP-1
Il est également possible d’associer un traitement à base d’insuline basale avec un traitement à base de GLP-1 RA. Il existe pour cela des combinaisons fixes permettant, en une seule injection quotidienne, de combiner ces deux traitements.
Les combinaisons disponibles combinent l’effet de l’insuline basale avec celui du GLP-1 RA, qui agit soit sur la glycémie à jeun, soit sur la glycémie postprandiale.