Why choose PRALUENT®?
The usual starting dose for PRALUENT is 75 mg administered subcutaneously once every 2 weeks. Patients requiring larger LDL-C reduction (>60%) may be started on 150 mg once every 2 weeks, or 300 mg once every 4 weeks (monthly), administered subcutaneously.2



Intensive, fast, and sustained LDL-C reduction1,2¥
Demonstrated to get patients to LDL-C goals in a post hoc analysis4§
The only LLT beyond statins associated with a reduction in all-cause mortality in a CVOT1,2‖
Established, long-term safety profile2
Unique one-monthly pen for your patients requiring >60% LDL-C reduction2,3¶
- New Next-gen PRALUENT pen now available2
The long-term efficacy and safety of PRALUENT has been evaluated in an extensive clinical trial programme:
The ODYSSEY OUTCOMES trial was a CVOT with ~19,000 very high-CV-risk ACS patients, over 5 years1,2,5,6**
Background: ODYSSEY OUTCOMES aimed to determine whether PRALUENT would improve cardiovascular outcomes after an acute coronary syndrome in patients receiving high-intensity statin therapy.1
Multicentre, double-blind, placebo-controlled, phase 3 trial** N=18,9241
PRALUENT SC Q2W + max tolerated statins (n=9,462)1
PLACEBO SC Q2W + max tolerated statins (n=9,462)1
Primary endpoint:
MACE1
If needed, dose adjustment occurred at month 2 visit1
In ODYSSEY OUTCOMES:1
- Patients with very-high-risk recurrent CV were enrolled, and 100% of patients had an MI or unstable angina
- Patients received PRALUENT alongside the optimal standard of care - 89% were on high-intensity statins
- 2.8 years median follow-up, with over 8,000 patients (44%) eligible to be followed for 3–5 years
Primary endpoint: MACE
At 2.8 years (median follow up)1,2
CVOT
15% RRR
HR 0.85 (95% CI 0.78, 0.93)
P=0.0003
ARR 2.0%
NNT=49
Patients eligible for ≥3 years follow-up††7
Post hoc analysis
17% RRR
HR 0.83 (95% CI 0.74, 0.94)
P=0.003

ODYSSEY OUTCOMES is the longest randomised, placebo-controlled comparison of PCKS9 inhibition to date.1
All primary and secondary end points were adjudicated by physicians who were unaware of the trial-group assignments.1
To adjust for multiplicity, the results of the main secondary end points were tested in hierarchical fashion if the risk of the composite primary end point was found to be significantly lower in the PRALUENT group than in the placebo group.1
PRALUENT demonstrated powerful LDL-C reduction vs placebo1,2§§

The LDL-C reduction achieved by PRALUENT was proven to be:
Intensive
>62% LDL-C reduction relative to placebo at 4 months1§§
Fast
4 weeks to reach maximum effect2‖‖
Sustained
4 years of LDL-C reduction1¶¶
LDL-C reduction within ODYSSEY outcomes: on-treatment analysis1
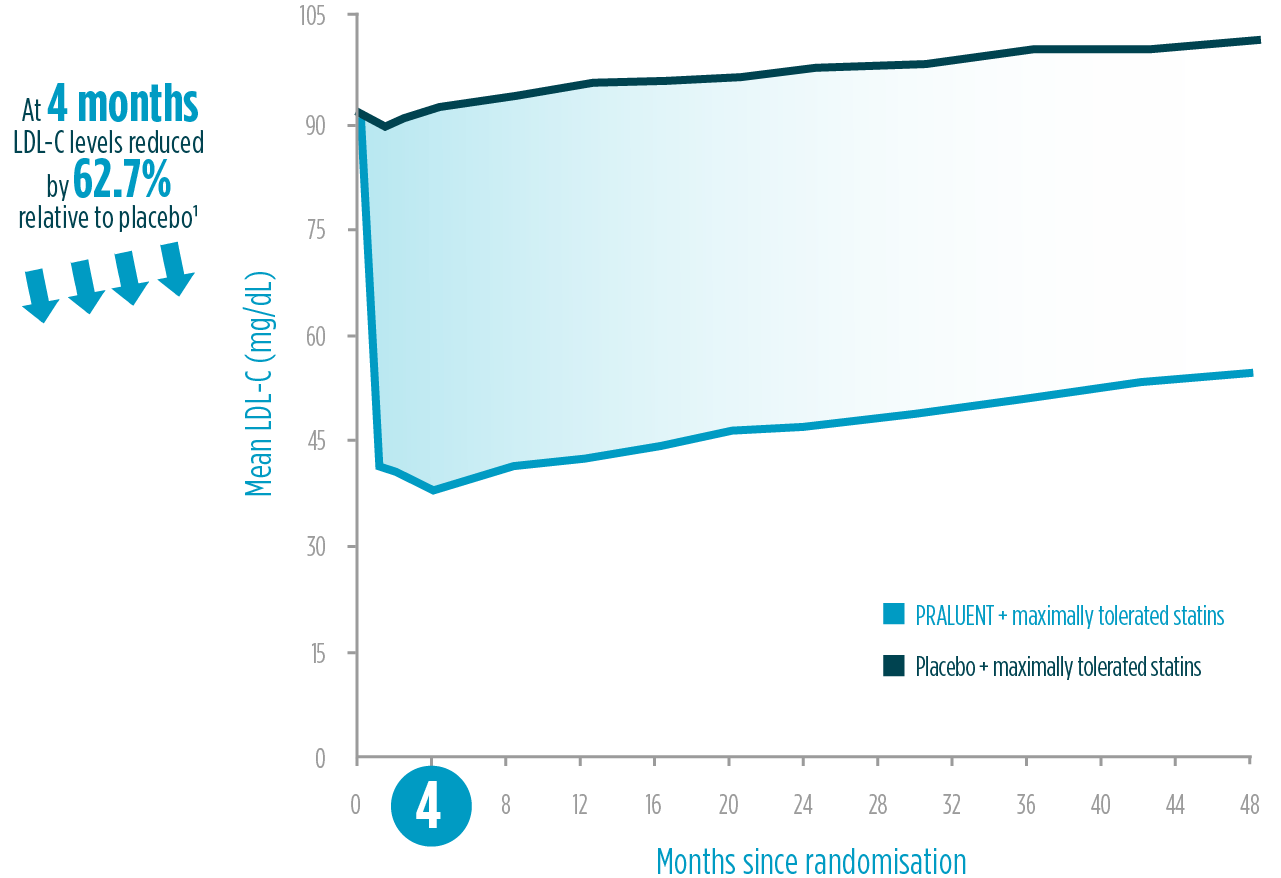
Adapted from Schwartz et al. 2018.1
Making PRALUENT an option for your ACS patients who are at immediate risk of another CV event and need LDL-C reduction that is both fast and sustained.8,9
Reaching LDL-C goal
In patients at very-high-risk and with an LDL-C goal of <55 mg/dL and a reduction of ≥50% from baseline:5

95% of ACS patients reached ≤55 mg/dL LDL-C goal with PRALUENT4***
in a post hoc assessment4***
All-cause mortality
The ODYSSEY OUTCOMES trial also assessed all-cause mortality reduction in patients treated with PRALUENT vs placebo:1,2,10§¶¶
Overall population‡‡‡
15% RRR
HR 0.85 (95% CI 0.73, 0.98)
P=0.0261§
Continued risk reduction over time§§
After 1 year¥¥¥
21% RRR
HR 0.79 (95% CI 0.66, 0.94)
P=0.0073
In a post hoc analysis
After ≥3 years¥¥¥,‡‡‡
22% RRR
HR 0.78 (95% CI 0.65, 0.94)
P=0.01
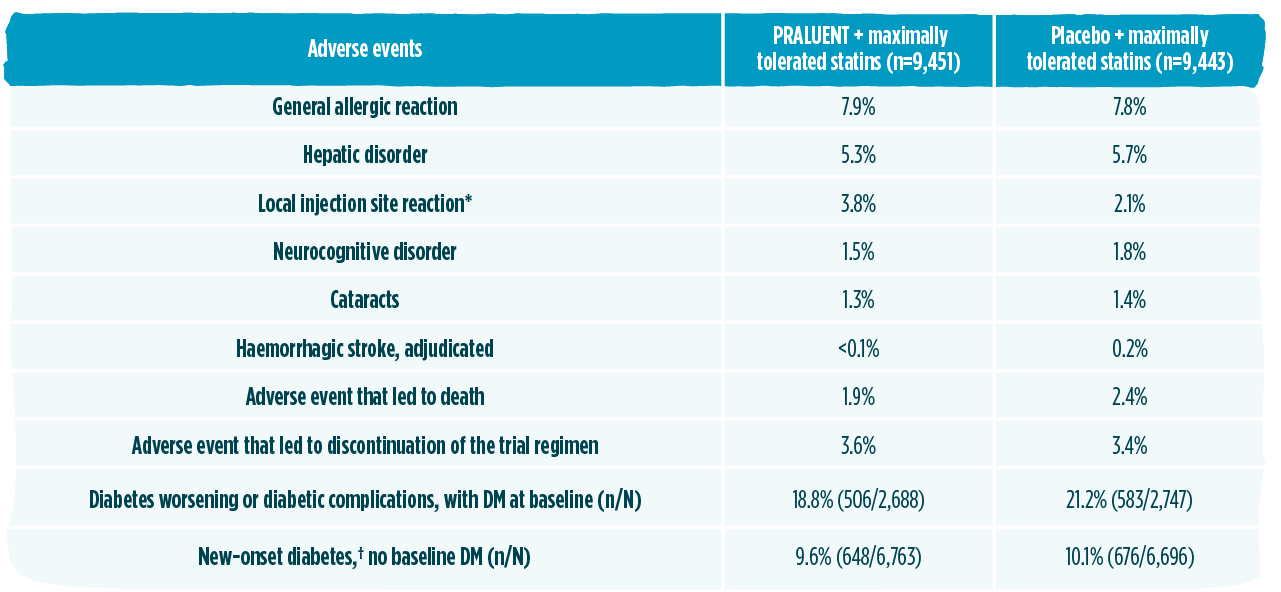
PRALUENT has established long-term safety profile and experience1,2,7,11
The safety profile in ODYSSEY OUTCOMES was comparable to placebo and consistent with the overall safety profile described in the phase 3 controlled trials1,2
Up to 5 years long-term safety – the longest randomised, placebo-controlled comparison of PCSK9 inhibition to date7‖‖‖
In 2023, approximately 570,000 patients globally have been treated with PRALUENT11§§§
*P<0.001.1
†New-onset diabetes was defined according to the presence of 1 or more of the following, with confirmation of the diagnosis by blinded external review by experts in the field of diabetes: an adverse event report, a new prescription for diabetes medication, a glycated haemoglobin level of at least 6.5% on 2 occasions (and a baseline level of <6.5%), or a fasting serum glucose level of at least 126 mg/dL (7.0 mmol/L) on 2 occasions (and a baseline level of <126 mg/dL).1
The safety profile in ODYSSEY OUTCOMES was consistent with the overall safety profile described in the phase 3 controlled trials. The only adverse reaction in ODYSSEY OUTCOMES occurring with higher incidence compared to placed was injection site reaction (P<0.001).1,2
PRALUENT: the only PCSK9i with the option of a once-monthly single injection2
The usability and LDL-C reduction of the PRALUENT pen was assessed in the SYDNEY Device trial, and the following information is related to the ease-of-use associated with the PRALUENT pen.3
PRALUENT reaffirmed rapid and sustained LDL-C reduction with the once-monthly pen3
SYDNEY Device (once-monthly pen) Study:
A 16-week multicentre, randomised, open-label study in the US with 69 hypercholesterolaemia patients despite receiving statin with or without other lipid-lowering therapy randomly received supervised, self-administered PRALUENT.3
Patients were randomised to receive either:
- Self-administered SYDNEY device 1 x 300 mg injection (n=35) or auto injector 2 x 150 mg (n=34)3
- The primary endpoint was PTC, which was measured at a rate of 0.5% (n=1) (95% CI, 0.0%–3.2%) during unsupervised injections and was not related to the device3****
For their first dose, patients received supervised, self-administered alirocumab 300 mg via 1 x 300 mg injection with the SYDNEY device (n=35) or 2 x 150 mg injections with the currently approved AI (n=34). All continuing patients subsequently received unsupervised, self-administered alirocumab 300 mg Q4W using the SYDNEY device at Weeks 4, 8, and 12.3
Mean LDL-C reduction from baseline at week 4 was 66.2% with the SYDNEY device3
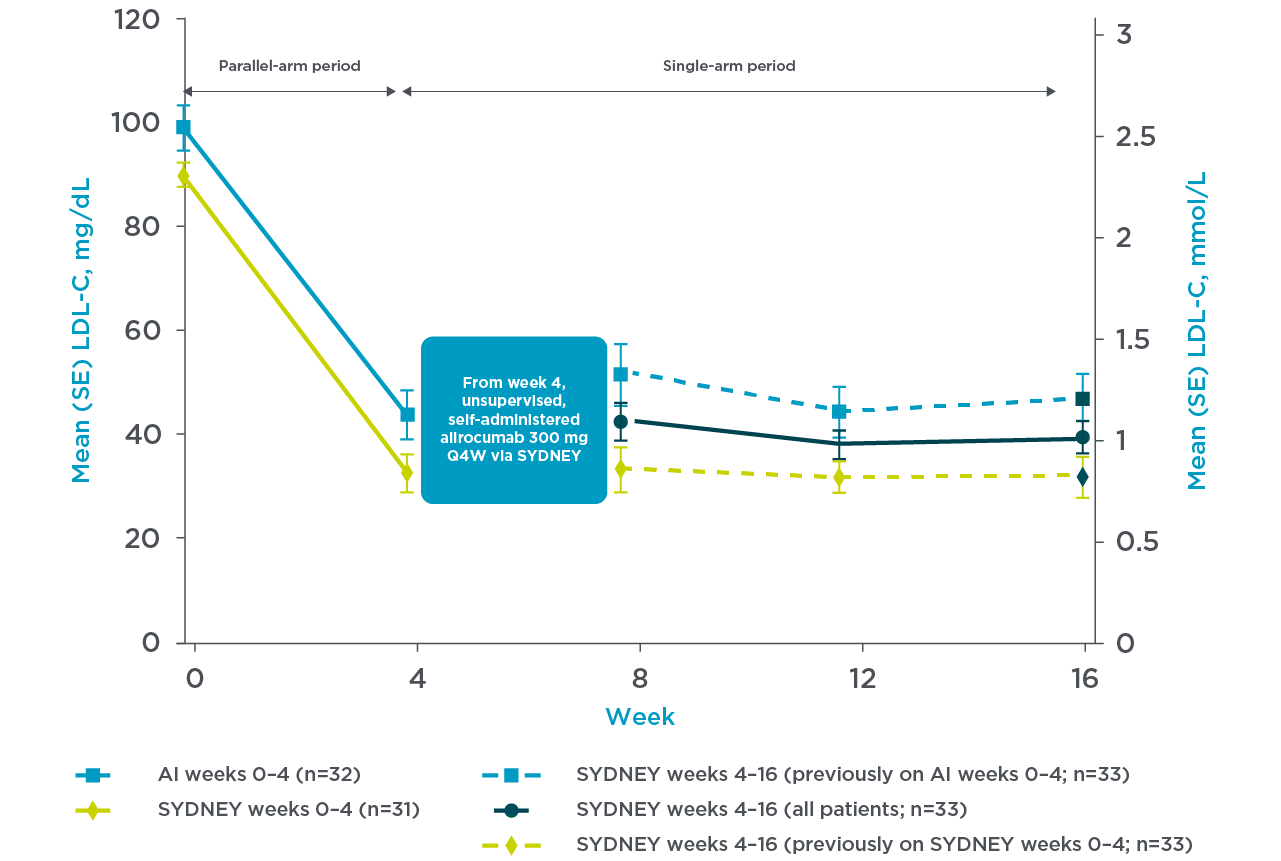
Adapted from Frias et al. 2020.3
PRALUENT self-administration has been optimised vs. previous devices.2
Now, all doses are injected with one device - the new Next-gen PRALUENT pen.
Streamlined 2-step operation
'remove the cap and inject'
Buttonless activation
Hidden needle in a secure protective cover
Convenient self-administration available across all doses
The Next-gen PRALUENT pen has designed to help patients self-inject confidently and correctly
More information
Click a link below to learn more about PRALUENT
What is PRALUENT?
Click here to learn about how PRALUENT works and who it is for
How to use PRALUENT?
Click here to learn more about how PRALUENT should be used
Indication, Dosing, and Footnotes
Primary hypercholesterolaemia and mixed dyslipidaemia2
PRALUENT® is indicated in adults with primary hypercholesterolaemia (heterozygous familial and non-familial) or mixed dyslipidaemia, and in paediatric patients 8 years of age and older with heterozygous familial hypercholesterolaemia (HeFH) as an adjunct to diet:2
- in combination with a statin or statin with other lipid lowering therapies in patients unable to reach LDL-C goals with the maximum tolerated dose of a statin or,
- alone or in combination with other lipid-lowering therapies in patients who are statin-intolerant, or for whom a statin is contraindicated.
Established atherosclerotic cardiovascular disease2
PRALUENT® is indicated in adults with established atherosclerotic cardiovascular disease to reduce cardiovascular risk by lowering LDL-C levels, as an adjunct to correction of other risk factors:
- in combination with the maximum tolerated dose of a statin with or without other lipid-lowering therapies or,
- alone or in combination with other lipid-lowering therapies in patients who are statin-intolerant, or for whom a statin is contraindicated.
Adults2
Prior to initiating PRALUENT® secondary causes of hyperlipidaemia or mixed dyslipidaemia (e.g., nephrotic syndrome, hypothyroidism) should be excluded.
The usual starting dose for alirocumab is 75 mg administered subcutaneously once every 2 weeks. Patients requiring larger LDL-C reduction (>60%) may be started on 150 mg once every 2 weeks, or 300 mg once every 4 weeks (monthly), administered subcutaneously.
The dose of PRALUENT® can be individualised based on patient characteristics such as baseline LDL-C level, goal of therapy, and response. Lipid levels can be assessed 4 to 8 weeks after treatment initiation or titration, and dose adjusted accordingly (up-titration or down-titration).
If additional LDL-C reduction is needed in patients treated with 75 mg once every 2 weeks or 300 mg once every 4 weeks (monthly), the dosage may be adjusted to the maximum dosage of 150 mg once every 2 weeks.
HeFH in paediatric patients 8 years of age and older:2
| Body weight of patients | Recommended dose | Recommended dose if additional LDL-C reduction is needed* |
| Less than 50 kg | 150 mg once every 4 weeks | 75 mg once every 2 weeks |
| 50 kg or more | 300 mg once every 4 weeks | 150 mg once every 2 weeks |
Adapted from PRALUENT® Summary of Product Characteristics. 2023.2
* Lipid levels can be assessed 8 weeks after treatment intiation or titration and dose adjusted accordingly.2
If a dose is missed, the dose should be administered as soon as possible and thereafter, dosing should be resumed on the original schedule.
*62.7% LDL-C reduction compared to placebo at 4 months in ODYSSEY OUTCOMES trial.1
†MACE: primary composite endpoint of CHD death, nonfatal myocardial infarction, fatal and nonfatal ischaemic stroke, or unstable angina requiring hospitalisation. HR 0.85 (95% CI 0.78, 0.93), P=0.0003.1,2
‡With only nominal statistical significance by hierarchical testing; HR 0.85 (95% Cl 0.73, 0.98), P=0.0261.1,2
¥PRALUENT significantly reduced risk of MACE (primary endpoint) in the overall trial population (N=18,924) in the ODYSSEY OUTCOMES trial (15% RRR, HR 0.85 (95% CI 0.78, 0.93), P=0.0003) and was associated with a reduction in all-cause mortality (15% RRR, HR 0.85 (95% CI 0.73, 0.98), P=0.0261) with only nominal statistical significance by hierarchical testing. 62.7% LDL-C reduction compared to placebo at 4 months in ODYSSEY OUTCOMES trial. 4 weeks to reach maximal effect based on a summary of ten phase 3 trials (five placebo-controlled, five ezetimibe-controlled) in high and very high-CV-risk patients. LDL-C reduction was sustained at 54.5% relative to placebo at 4 years in patients following an ACS event in the ODYSSEY OUTCOMES trial.1,2
§A post hoc assessment using data from the ODYSSEY OUTCOMES trial. With PRALUENT, 94.6% of patients achieved LDL-C,1.4 mmol/L at ≥1 post-baseline measurement vs. 17.3% with placebo.4
‖In a CVOT, with only nominal statistical significance by hierarchical testing (HR 0.85, 95% CI 0.73, 0.98), P=0.0261.2
¶For patients requiring LDL-C reduction >60%, PRALUENT is the only PCSK9i with once-monthly single injection in a pre-filled pen.2
**Patients were randomised for 1 to 12 months after an acute coronary syndrome and had an LDL-C ≥70 mg/dL or non-HDL-C ≥100 mg/dL, or an ApoB of ≥80 mg/dL.1
††Of 8,242 patients (43.5%) eligible for 3 to 5 years’ follow-up, 8,228 received ≥1 dose of PRALUENT, comprising 24,610 patient-years of observation, with a median follow-up of 3.3 years; 6,651 patients were eligible for 3 to 4 years’ follow-up, and 1,574 patients were eligible for 4 to 5 years’ follow-up.7
‡‡Death from CHD, non-fatal MI, unstable angina requiring hospitalisation, or an ischaemia-driven coronary revascularisation procedure.1
¥¥Death from cardiovascular causes, non-fatal MI, unstable angina requiring hospitalisation, an ischaemia-driven coronary revascularisation procedure, or non-fatal ischaemic stroke.1
§§62.7% LDL-C reduction compared to placebo at 4 months in ODYSSEY OUTCOMES trial.1
‖‖Based on a summary of ten phase 3 trials (five placebo-controlled, five ezetimibe-controlled) in high and very high CV-risk patients.2
¶¶In patients following an ACS event in the ODYSSEY OUTCOMES trial.2
***95% of ACS patients reached <55 mg/dL LDL-C goal with PRALUENT, in addition to maximally tolerated statins vs 17% in the control placebo group.4
†††Intention-to-treat population during the overall trial period10
‡‡‡From a sub-analysis of mortality data.10
¥¥¥Prespecified analysis.10
§§§During the first year: HR 1.01 (95% CI 0.77, 1.32); for patients eligible for <3 years of follow-up: HR 0.96 (95% CI 0.76, 1.21).10
‖‖‖A post hoc analysis of ODYSSEY OUTCOMES, to describe the efficacy and safety of PRALUENT in a pre-specified subgroup of patients eligible for a follow-up of 3–5 years. 8,242 patients were eligible for >3 years follow-up. 8,242 patients were eligible for >3 years follow-up. 6,651 patients were eligible for 3–4 years, and 1,574 patients were eligible for 4–5 years follow-up, comprising 24,610 patient-years of observation. Of 8,242 patients, 8,228 received at least one dose of PRALUENT. The safety of PRALUENT was similar to placebo except for an excess of local injection site reactions.7
¶¶¶Excluding the USA, based on estimation of IQVIA MAT December 2023 data.11
****A multicentre, randomised, open-label, 16-week study in the United States. For their first dose, 69 patients with hypercholesterolaemia despite receiving statin with or without other lipid-lowering therapy randomly received supervised, self-administered alirocumab 300 mg via 1 × 300 mg injection with the SYDNEY device (n=35) or 2 x 150 mg injections with the currently approved AI (n=34). All continuing patients subsequently received unsupervised, self-administered alirocumab 300 mg Q4W using the SYDNEY device at weeks 4, 8, and 12. PTC happened at W4 and was classified as patient related. No PTCs were reported during supervised injections.3
††††ODYSSEY APPRISE is a prospective, single-arm, phase 3b open-label study of PRALUENT in Europe and Canada. This post hoc analysis of the ODYSSEY APPRISE study examined patient adherence to treatment, and efficacy and safety of PRALUENT according to background statin therapy and prior ezetimibe medication, in patients with severe hypercholesterolaemia at high or very high risk of future CV events. Patients received PRALUENT 75 or 150 mg every 2 weeks.12
ACE = angiotensin-converting enzyme; ACS = acute coronary syndrome; AI = autoinjector; ApoB = apolipoprotein B; ARB = angiotensin II receptor blocker; ARR = absolute risk reduction; CHD = cardiovascular heart disease; CI = confidence interval; CV = cardiovascular; CVOT = cardiovascular outcomes trial; HDL-C = high-density lipoprotein cholesterol; HeFH = heterozygous familial hypercholesterolaemia; HR = hazard ratio; LDL-C = low-density lipoprotein cholesterol; LLT = lipid-lowering therapy; MACE = major adverse cardiovascular event; MI = myocardial infarction; NNT = number needed to treat; PCSK9 = proprotein convertase subtilisin/kexin type 9; PCSK9i = proprotein convertase subtilisin/kexin type 9 inhibitor; PTC = product technical complaint; Q2W = every 2 weeks; Q4W = every 4 weeks; RRR = relative risk reduction; SC = subcutaneous; TEAE = treatment emergent adverse event.
-
Schwartz GG, Steg PG, Szarek M, et al. N Engl J Med. 2018;379(22):2097–2107.a
-
PRALUENT (alirocumab) Summary of Product Characteristics. Paris, France: sanofi-aventis groupe; December 2023.
-
Frias JP, Koren MJ, Loizeau V, et al. Clin Ther. 2020;42(1):94–107.
-
Landmesser U, McGinniss J, Steg PG, et al. Eur J Prev Cardiol. 2022;29(14):1842–1851.
-
Mach F, Baigent C, Catapano AL, et al. Eur Heart J. 2020;41(1):111–188.
-
Schwartz GG, Steg PG, Szarek M, et al. N Engl J Med. 2018;379(22):2097–2107. Supplementary Appendix.
-
Goodman SG, Steg PG, Poulouin Y et al. Long-term Efficacy, Safety, and Tolerability of Alirocumab in 8,242 Patients Eligible for 3 to 5 Years of Placebo-Controlled Observation in the ODYSSEY OUTCOMES Trial. J Am Heart Assoc. 2023;12(18):e029216. doi:10.1161/JAHA.122.029216.
-
Jernberg T, Hasvold P, Henriksson M, et al. Eur Heart J. 2015;36(19):1163–1170.
-
Chi G, Lee JJ, Kazmi SHA, et al. Clin Cardiol. 2022;45(3):299–307.
-
Steg PG, Szarek M, Bhatt DL, et al. Circulation. 2019;140(2):103–112.
-
Number of patients calculated and based on internal data. MAT December 2023
-
Banach M, Lopez-Sendon JL, Averna M, et al. Arch Med Sci. 2021;18(2):285–292.
-
Gaudet D, Lopez-Sendon JL, Averna M, et al. Eur J Prev Cardiol. 2022;28(17):1864–1872
For the development of the Sanofi-“Reflex”-autoinjector-platform (model series of NextGenerationPraluent®Pen) human-factor-studies/tests with test persons/probands have been conducted, which assessed the pen within the scope of the development and optimization process. These user tests referred exclusively to the handling of the autoinjector. Data on file, 2017, 2019.
Articles liés


Prevent the Event
Despite major advances in our understanding of atherosclerosis, ASCVD remains the leading cause of death globally.1

Lipid management in patients with diabetes mellitus
Key principles of lipid management (LDL-C in particular) in accordance with the 2019 ESC/EAS Guidelines for the management of dyslipidaemias, and the 2023 ESC guidelines for the management of cardiovascular disease in patients with diabetes.
Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.