Fabry disease
Fabry disease is an X-linked lysosomal storage disease due to a defect in the gene encoding the lysosomal enzyme alpha-galactosidase A (α-Gal A), causing progressive cellular accumulation of the substrate globotriaosylceramide (GL-3) and globo-triaosylsphingosine (lyso-GL-3).

Fabry disease is an X-linked lysosomal storage disease due to a defect in the gene encoding the lysosomal enzyme alpha-galactosidase A (α-Gal A), causing progressive cellular accumulation of the substrate globotriaosylceramide (GL-3) and globo-triaosylsphingosine (lyso-GL-3).
This accumulation occurs in a variety of cell types and can lead to debilitating symptoms such as neurological pain, angiokeratoma, hypohidrosis in childhood, in girls usually a few years later than in boys. With age, progressive damage to the vital organs develops in both sexes that leads to organ failure. End-stage kidney disease and life-threatening cardiovascular or cerebrovascular complications limit life expectancy.
Although the disease is X-linked, most women develop symptoms. Fabry disease is pan-ethnic. Newborn screenings report frequencies of 1 in 22,570 men for the classic phenotype and of 1 in 1,390 men for the late-onset phenotype.1-3
Fabry disease is classified into two main phenotypes:1,3,4
- Classic – absent of very low α-GAL A activity, multiple-organ systems involved, presentation generally begins in childhood
- Nonclassic – also referred to as late-onset, varying levels of residual α-GAL A activity and symptoms are more variable, most frequently beginning in adulthood
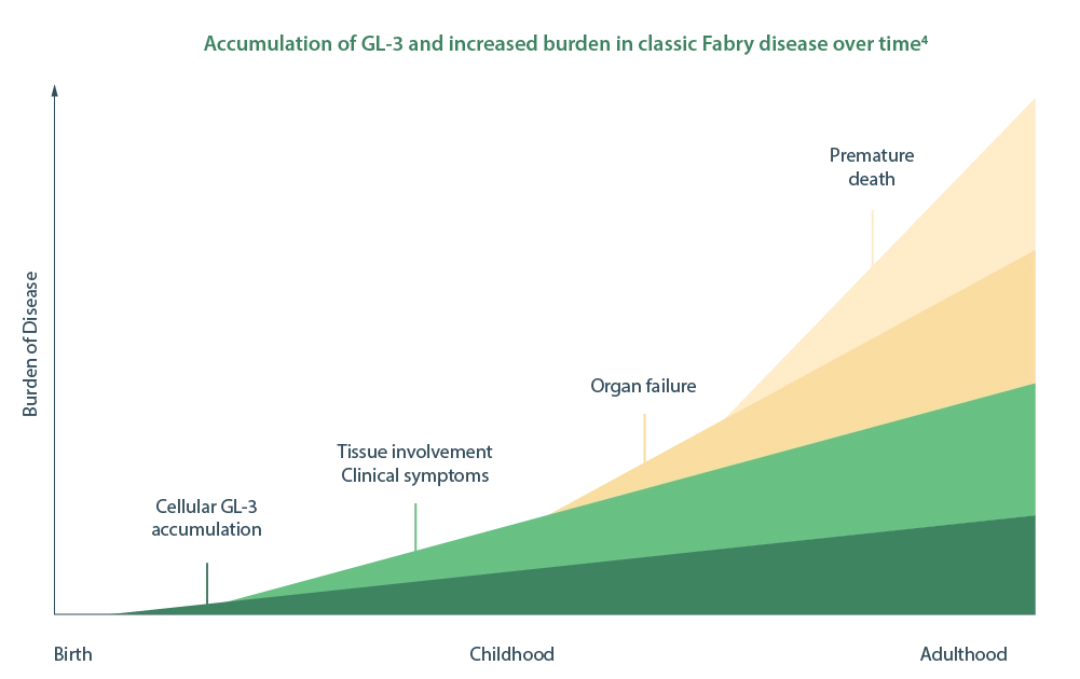

α-GAL A, α-galactosidase A; GLA, galactosidase alpha; GL-3, globotriaosylceramide.
Irreversible damage to multiple vital organs can cause renal, cardiovascular, and cerebrovascular complications if Fabry disease is left untreated.1,3,6
Multisystemic signs and symptoms
- Neuropathic pain
- Pain crises
- Heat and/or cold intolerance
- Hypohidrosis/anhidrosis
- Hearing loss/tinnitus
- Dizziness
- Burning of hands and feet
- Angiokeratomas
- Nausea/vomiting
- Diarrhea and constipation
- Abdominal pain and/or bloating
- Difficulty gaining weight in childhood
- Cornea verticillate
- Tortuous vessels (conjunctival)
- Fabry cataract
- Corneal whorling
- Aortic stiffness
- Depression/anxiety
- Fatigue
- Dyspnea
- Wheezing
- Chronic cough
- Shortness of breath
- Progressive LVH
- Chest pain
- Bradycardia
- Cardiomyopathy
- Arrhythmias, some of which can be lethal
- Ventricular fibrosis
- Heart failure
- Pathological albuminuria/proteinuria
- Decreased glomerular filtration rate
- Kidney failure
- Transient ischemic attack
- Early stroke
Patients with Fabry disease experience an approximated 16-year reduction in lifespan for males and a 5- to 14-year reduction for females compared with the general population14-16

-
Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30.
-
Germain D., Fabry Disease. Orphanet encyclopedia, March 2022, http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=324
-
Ortiz A, Germain DP, Desnick RJ et al. Fabry disease revisited: management and treatment recommendations for adult patients. Molecular genetics and metabolism 123.4 (2018): 416-427.
-
Arends M et al. Mol Genet Metab. 2017;121:157-161.
-
Eng CM et al. J Inherit Metab Dis. 2007;30(2):184-192.
-
Wanner C et al. Mol Genet Metab. 2019;126(3):210-211.
-
Lidove O et al. Intl J Clin Pract. 2006;60(9):1053-1059.
-
Burlina A et al. BMC Neurol. 2011;11(61).
-
Sodi A et al. Br J Ophthalmol. 2007;91(2):210-214.
-
Zarate Y, Hopkin R. Lancet. 2008;372:1427-1435
-
Yousef Z et al. Eur Heart J. 2013;34(11):802-808
-
Linhart A et al. J Inherit Metab Dis. 2001;24(2):75-83.
-
Shi Q et al. J Stroke Cerebrovasc Dis. 2014;25(5):985-992.
-
Waldek S et al. Genet Med. 2009;11(11):790-796.
-
Mehta A et al. J Med Genet. 2009;46(8):548-552.
-
Arias E et al. NVSS Vital Statistics Rapid Release Report No. 015. 2021;1-12. Available at: https://www.researchgate.net/publication/362689060_Provisional_Life_ Expecta ncy_Estimates_for_2020. Accessed: May 2024.



Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.