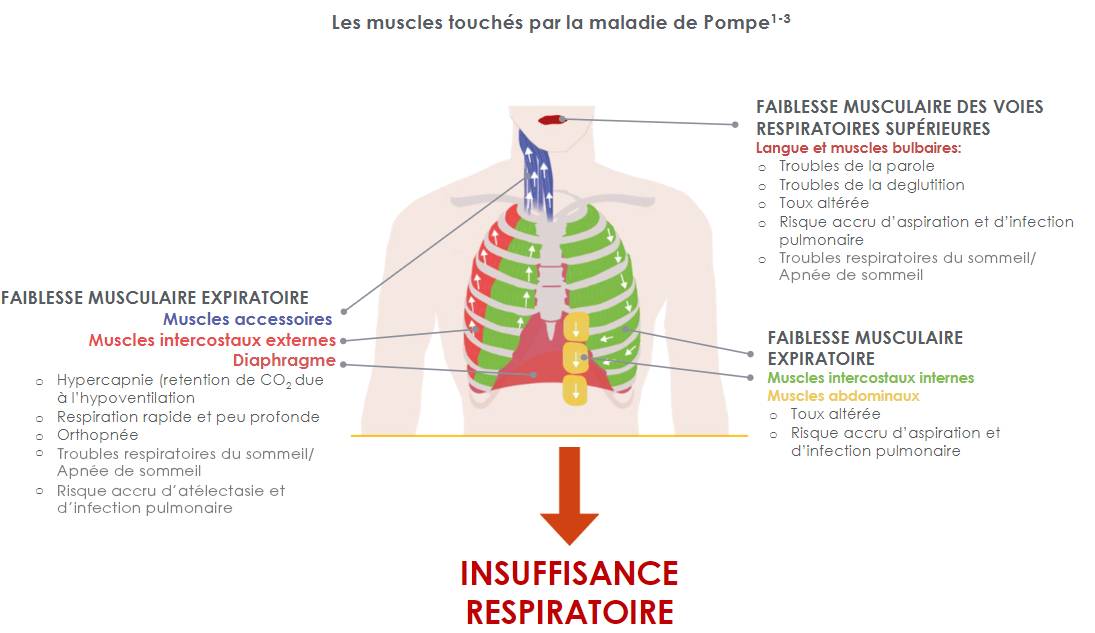
Atteinte respiratoire de la forme tardive de la maladie de Pompe (LOPD)

Une maladie musculaire aux implications respiratoires majeures
Les principaux symptômes respiratoires :
Dans la forme tardive de la maladie de Pompe (LOPD), une faiblesse des muscles respiratoires provoque la dysfonction respiratoire.1-3
%20(1).jpg)
L’atteinte respiratoire de la maladie de Pompe est un signe important de l'atteinte musculaire, mais il est trop souvent éclipsé par la perte de mobilité.4
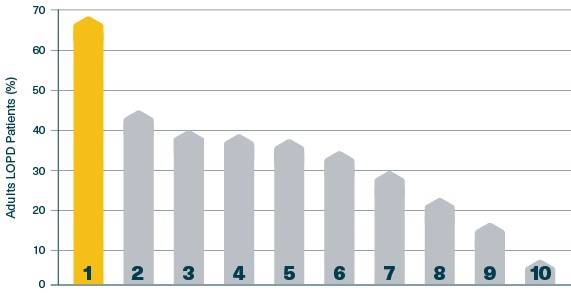
Classement de la fréquence des symptômes5 :
%20(1).jpg)
1. Essouflement après l’exercice
2. Assistance respiratoire requise
3. Ventilation non invasive requise
4. Troubles du sommeil/Apnée de sommeil
5. Orthopnée
6. Essoufflement au repos
7. Détresse respiratoire
8. Pneumonie
9. Supplément/oxygène requis
10. Ventilation invasive requise
Un retard de diagnostic subsiste et engendre des conséquences majeures pour les patients.6,7
Les patients atteints de la forme tardive de la maladie de Pompe peuvent attendre 6 à 13 ans entre l’apparition de leurs premiers symptômes et le diagnostic.6,7
Or, on sait qu’en absence de traitement chez ces patients, la probabilité qu’ils aient besoin:
- d’un fauteuil roulant augmente, en moyenne, de 13 % par an
- d’une assistance respiratoire augmente, en moyenne, de 8% par an
On sait également qu’en absence de traitement, l’âge médian au moment du décès d’un patient LOPD est de 54 ans.5,8
Les difficultés à respirer apparaissent fréquemment et à un stade précoce de la maladie.
![]()
L’atteinte respiratoire chez les patients LOPD: Facts & Figures
Tous les patients finissent par présenter un dysfonctionnement respiratoire.9
La plupart des patients sont affectés par une faiblesse des muscles respiratoires:
Environ 60 % des patients LOPD ont une réduction de la capacité vitale forcée (CVF) <80 % de la valeur prédite, et 30 à 40 % ont une réduction <60 % de la valeur prédite.1
La CVF en position couchée diminue de 5,5 % par an en l’absence de traitement.10
Plus de 70% des patients non traités développent une insuffisance respiratoire chronique.11
Enfin, l'insuffisance respiratoire est la cause de décès la plus fréquente.12
La faiblesse progressive du diaphragme est la principale cause de dysfonctionnement respiratoire13
Notez l'augmentation limitée de la longueur verticale chez le patient Pompe par rapport à l'augmentation chez le volontaire sain.13
IRM à l'inspiration maximale
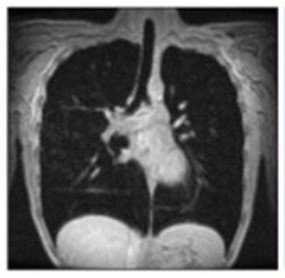 Volontaire sain
Volontaire sain
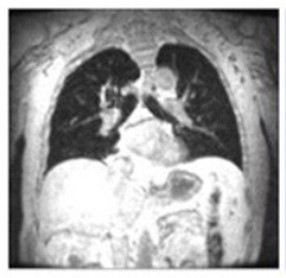 Patient Pompe
Patient Pompe
La faiblesse du diaphragme altère son mouvement et sa fonction, et peut évoluer jusqu’à sa paralysie.14
Elle se manifeste initialement pendant le sommeil en raison de l’effet de la gravité en position couchée sur le dos.1,13-14 Dans une certaine mesure, la fonction altérée du diaphragme peut être compensée par les muscles respiratoires intercostaux, accessoires et abdominaux.15

- Insuffisance respiratoire restrictive22
Des études récentes montrent une forte prévalence de la maladie de Pompe chez les patients présentant initialement une insuffisance respiratoire.16-17
Par exemple, dans une étude 1,4% des patients pour lesquels il y avait une suspicion de trouble neuromusculaire avec atteinte respiratoire ont été testés positifs à la maladie de Pompe (n=140).16
Dans une autre étude incluant des patients atteints de paralysie diaphragmatique d'étiologie inconnue (n-18), ce pourcentage s’élevait à 16,7%.17
- Faiblesse proximale/axiale23
Avec une faiblesse des muscles proximaux, les activités quotidiennes deviennent un défi pour les patients.10
80 % des patients LOPD déclarent avoir des problèmes avec les tâches quotidiennes en raison de la perte de force et de fonction musculaires.18
Par exemple, au moment du diagnostic, 50 % des patients ont des difficultés à se lever d'une chaise, près de la moitié éprouvent des difficultés pendant le travail ou les études, et > 20 % sont dépendants d'un fauteuil roulant ou d'une assistance respiratoire.18
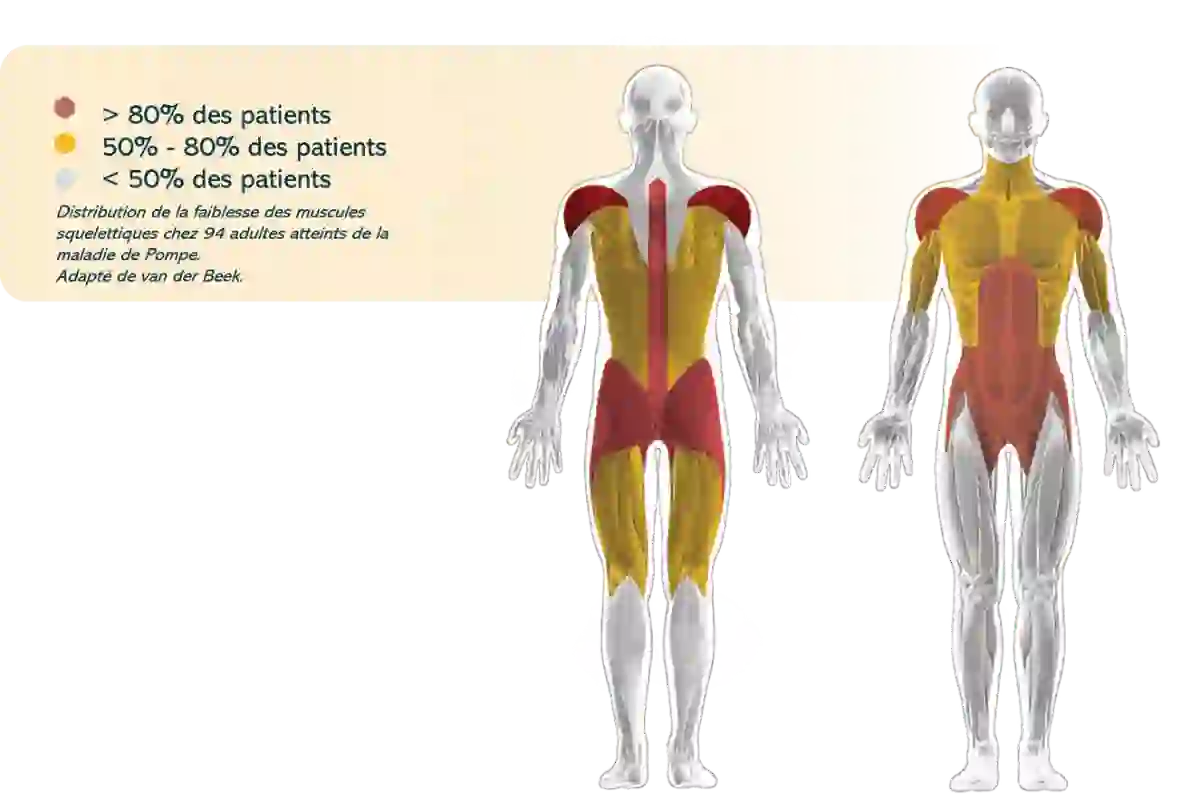
Les muscles proximaux des membres inférieurs sont généralement plus sévèrement touchés que les muscles proximaux des membres supérieurs24
Les muscles du tronc, paraspinaux et abdominaux sont souvent touchés précocement dans l’évolution de la maladie5
- HyperCKémie asymptomatique22

9/10 patients LOPD ont un taux sérique de créatine kinase élevé en raison de la souffrance musculaire.6,19
Les valeurs de CK sont souvent légèrement à modérément élevées : 1 à 1,5 fois la limite supérieure de la normale.21
Les valeurs de CK normales n'excluent pas nécessairement la maladie de Pompe.21
Des taux élevés de CK sont également plus fréquemment observés avec des enzymes hépatiques élevées : alanine transaminase (ALT), aspartate transaminase (AST), lactate déshydrogénase (LDH)20
Le diagnostic se fait en 3 étapes :
- Mesure de l'activité enzymatique résiduelle de l’ α-glucosidase acide sur une un papier buvard DBS (Dry Blood Spot test).
- Si il y a un déficit de l’activité enzymatique : une analyse génétique est réalisée à partir du même DBS ou d’un tube de sang complet, pour confirmer la mutation du gène GAA
- Et enfin une confirmation du déficit enzymatique sur culture de fibroblastes est réalisée par le centre de référence neuromusculaire.

En savoir plus?
- J'aimerais en savoir plus sur les options de traitement de la maladie de Pompe
- J'aimerais recevoir un kit de diagnostic avec des cartes buvard DBS (Dried Blood Spot test) et des instructions d'utilisation, ainsi qu'un aperçu des centres de références belges
Pour toute autre question, vous pouvez contacter votre Product Specialist:
- etienne.delhoux@sanofi.com pour le sud est de la Wallonie et le Luxembourg
- marie.kruk@sanofi.com pour le sud ouest de la Wallonie et Bruxelles
- Kishnani PS et al. Genet Med 2006; 8(5): 267-88. 2.Perrin C et al. Muscle Nerve 2004; 29(1): 5-27. 3. van der Beek NA et al. Orphanet J Rare Dis 2012; 7(1): 88. 4. Cupler et al. Muscle Nerve 2012; 45(3): 319-333. 5. Byrne et al. Mol Genet Metab 2011; 103(1): 1-11. 6. Kishnani PS et al. On behalf of the Pompe Registry boards of advisors. Am J Med Genet. 2013; 161 A(10): 2431-2443. 7. Vanherpe P et al. Orphanet J Rare Dis 2020; 15: 83. 8. Hagemans et al. Neurology; 64: 2139-2141. 9. Raben et al. Curr Mol Med 2002; 2: 145-66. 10. Wokke JH et al. Muscle nerve et al. 2008; 38(4): 1236-45. 11. Boentert M et al. Int J Sci 2016; 17(10): E1735. 12. Winkel LPH et al. J Neurol 2005; 2525(80): 875-884. 13. Wens SC et al. BMC Pulm Med. 2015;15:54. 14. Newsom DJM et al. Q J Med 1976; 177: 87-100. 15. Mogalle K et al. PLoS ONE 2016; 11(7): e0158912. 16. Confalonieri M et al. Orphanet J Rare Dis 2019; 14(1): 62. 17. Guimaraes MJ et al. Rev Port Pneumol 2017; 23(4). 18. Rigter T et al. Mol Genet Metab 2012; 107(3): 449-455. 19. Toscano A, Schoser B J Neurol 2013; 260: 951-959. 20. Toscano et al. Ann Transl Med 2019; 7(13): 284. 21. AANEM. Muscle Nerver 2009; 40(1): 149-160. 22. Toscano et al. Acta Myol 2013; 32(2): 78-81. 23. Preisler et al. Mol Genet Metab 2013; 110(3): 287-9. 24. Reuser, Arnold J. J., et al. “Pompe Disease: Glycogen Storage Disease Type II, Acid alpha-glucosidase (Acid Maltase) Deficiency.” The OMMBID Eds. David L. Valle, et al. McGraw Hill, 2019


Les contenus accessibles par ce site sont destinés exclusivement aux Professionnels de Santé.
Les contenus accessibles par ce site sont destinés
exclusivement aux Professionnels de Santé.
Sanofi met a votre disposition, sur un portail unique dédié aux professionnels de santé, des contenus exclusifs comprenant des contenus scientifiques et des outils pour votre pratique quotidienne.