- Apresentação da doença
- Classificação da doença
- Progressão e prognóstico
- Apresentação clínica
- Genética e Epidemiologia
- Probabilidades de transmissão da doença de Gaucher
- Algoritmo de Diagnóstico
- Teste de diagnóstico e confirmação
- Rastreio familiar
- Tratamento da doença de Gaucher
- Monitorização periódica do doente de Gaucher
- O Registo de Gaucher do ICGG
- Article
- Source: Campus Sanofi
Apresentação da doença

Patologia subjacente
A doença de Gaucher é uma doença de sobrecarga lisossomal causada por uma variante patogénica no gene responsável pela produção da enzima β-glucosidase ácida. Já foram identificados mais de 300 alelos portadores de variantes patogénicas.1
A doença foi descrita pela primeira vez pelo estudante de medicina francês Phillipe Gaucher em 1882, ao observar uma jovem mulher com o baço aumentado e células ingurgitadas características.2 Pouco mais de 50 anos depois, Aghion relatou que pessoas com esta doença acumulavam um esfingolípido chamado glucosilceramida. Mas foi só em 1965 que Brady e colegas mostraram que a doença de Gaucher era causada pela redução da atividade da enzima β-glucosidase ácida.2
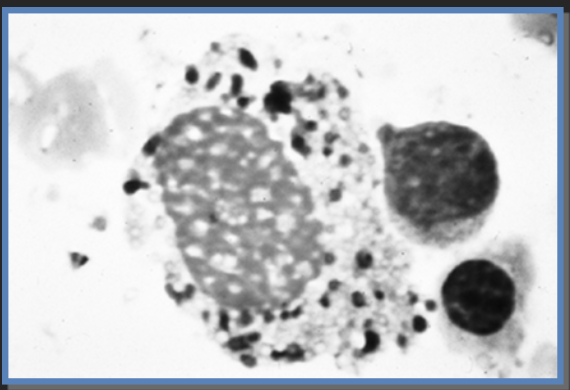
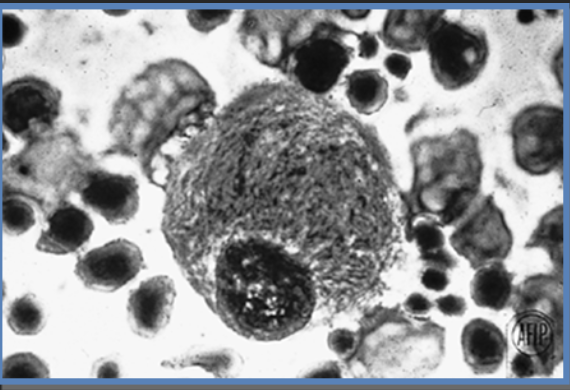
A enzima funcional tem atividade de decomposição de glucoesfingolípidos derivados da renovação fisiológica das membranas, particularmente das células sanguíneas. Variantes patogénicas no gene da β-glucosidase ácida desencadeiam a redução da atividade enzimática desta enzima, que depois se torna insuficiente para evitar a acumulação de um glucoesfingolípido, denominado glucosilceramida, nos lisossomas das células, principalmente nos macrófagos. Macrófagos com glucosilceramida acumulada, chamados células de Gaucher, acumulam-se nos órgãos. O armazenamento de células de Gaucher desencadeia a uma cascata de eventos fisiopatológicos, incluindo a produção de um estado inflamatório crónico e hipermetabólico.3
Esta é uma doença multissistémica que tem variações significativas nas suas manifestações clínicas, gravidade e curso. Uma deficiência parcial de β-glucosidase ácida está associada a doenças do fígado, baço, medula óssea e pulmão. Uma deficiência grave da enzima está também associada a manifestações neurológicas.4
Célula de Gaucher por acumulação de glucosilceramida (e outros substratos)
Macrófago normal
Sem acumulação nos lisossomas
Célula de Gaucher
Macrófago com glucosilceramida acumulada nos lisossomas
As imagens são uma cortesia do Prof. J.A. Barranger e de M. Judith Peterschmitt
- Mistry PK, Weinthal JA, Weinreb NJ. Disease state awareness in Gaucher disease: a Q&A expert roundtable discussion. Clin Adv Hematol Oncol. 2012;10:1-16.
- Brady RO. Gaucher’s disease: past, present and future. Bailleres Clin Haematol 1997;10(4):621-34.
- Grabowski G. Gaucher disease and other storage disorders. Hematology Am Soc Hematol Educ Program 2012;1:13-18 (doi: 10.1182/asheducation-2012.1.13)
- Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Bailleres Clin Haematol 1997;10(4):657-689.
Caminhar

Levantar-se-de-uma-cadeira

Subir-escadas

Levantar-os-braços-acima-da-cabeça

Vídeo-doença-de-Pompe2

Vídeo-Movimentos-de-Pompe---MARCHA_HCPs2

Vídeo-Movimentos-de-Pompe---SUBIR_ESCADAS_HCPs2

Vídeo-Movimentos-de-Pompe---SUBIR_ESCADAS_HCPs3

Vídeo-McIntosh-et-al-20202

Living-with-Pompe-disease-Sean-s-and-Cheryl-s-story

Testemunho-Shaylee-2
