
Enfermedad de Gaucher
Ciencia en Enfermedad de Gaucher
Descubra los contenidos destacados y manténgase al día con las últimas noticias.
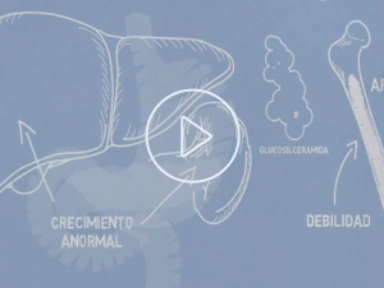
Qué es la enfermedad de Gaucher
La enfermedad de Gaucher es la enfermedad de depósito lisosomal de mayor prevalencia, causada por la deficiencia hereditaria de la enzima glucocerebrosidasa que conduce a la acumulación de glucocerebrósidos en los macrófagos de múltiples órganos. Su presentación clínica es heterogénea y puede incluir esplenomegalia, hepatomegalia, citopenias y afectación ósea de intensidad variable. El diagnóstico definitivo se establece mediante la medición de la actividad enzimática y la confirmación genética. Explore los recursos clínicos disponibles sobre la enfermedad de Gaucher.
¿Qué dicen los expertos?
Próximamente
Congresos y eventos
Inspírese y manténgase a la vanguardia de la innovación con los siguientes recursos:
Opciones terapéuticas

Mecanismo de acción en Gaucher
Explicación del mecanismo de acción molecular involucrado en la enfermedad de Gaucher y su impacto fisiopatológico.


-svg.2024-01-24-09-14-11.svg)


