Avaliações e Diagnóstico

Diagnóstico diferencial
A distinção da doença de Pompe de outras doenças é crucial para minimizar atrasos no diagnóstico e otimizar o prognóstico dos doentes1-4
Doentes com LOPD podem experienciar um atraso de 6 a 12,6 anos até receberem um diagnóstico definitivo e doentes com IOPD experienciam um atraso mediano de 1,4 meses desde o início dos sintomas até ao diagnóstico.1 O declínio clínico durante estes intervalos sem diagnóstico pode ser profundo, à medida que os danos irreversíveis e ameaçadores da vida progridem.2,5,6
O diagnóstico e tratamento atempados são essenciais para gerir os sintomas e melhorar o prognóstico dos doentes de Pompe. Suspeite de doença de Pompe em bebés, crianças e adultos, quando os sinais e sintomas sugerem degeneração muscular progressiva.2,3,7
Suspeite de doença de Pompe se vir algum dos seguintes sinais ou sintomas:
Em crianças e adultos com LOPD
- Fraqueza dos músculos proximais e/ou níveis de creatina quinase (CK, creatine kinase) moderadamente elevados e de forma persistente e sem explicação aparente2
Em lactentes com IOPD
- Cardiomegalia/cardiomiopatia, hipotonia, fraqueza rapidamente progressiva dos músculos esquelético, liso e ventilatório, dificuldades de alimentação e falha no atingimento de marcos de desenvolvimento2,8-10
Outras doenças podem ter sinais e sintomas similares aos da doença de Pompe. Inclua a doença de Pompe no seu diagnóstico diferencial, juntamente com outras doenças com apresentação semelhante2
Sobreposição de sintomas entre a LOPD e outras doenças mais comuns
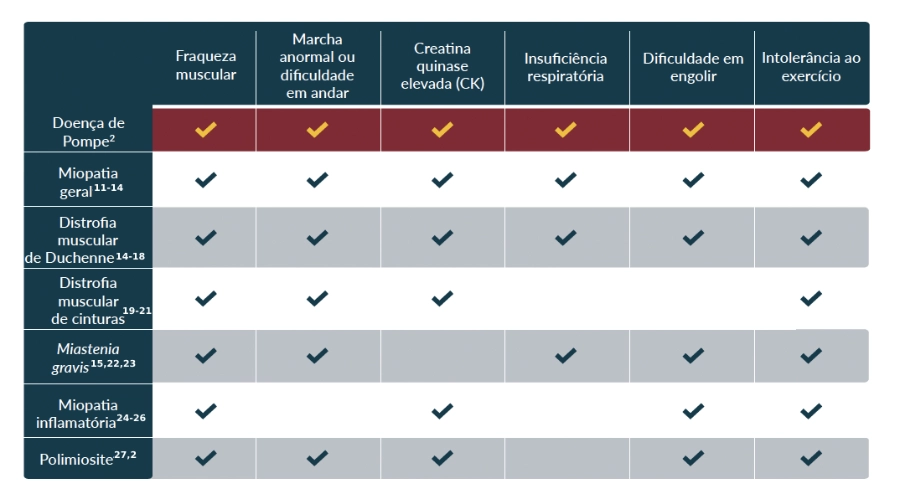
Sobreposição de sintomas entre a LOPD e outras doenças mais comuns2
| Doença | Sintomas partilhados com a IOPD | |
| Atrofia muscular espinhal (doença de Werdnig-Hoffman aguda) |
|
|
| Hipotiroidismo |
|
|
| Distrofia muscular congénita |
|
|
| Doença de Danon |
|
|
| Fibroelastose endocárdica |
|
|
| Deficiência de carnitina |
|
|
| Glicogenoses tipos III e IV |
|
|
| Cardiomiopatia hipertrófica idiopática |
|
|
| Miocardite |
|
|
| Distúrbios na cadeia mitocondrial/respiratória |
|
|
| Distúrbios peroxissomais |
|
|
Diagnóstico diferencial
Uma vez levantada a suspeita clínica, pode-se testar para a doença de Pompe medindo-se a atividade enzimática da α-glucosidase ácida (AGA)34
Considere testar para a doença de Pompe se observar:
- Fraqueza progressiva dos músculos proximais com ou sem insuficiência respiratória ou hiperCKemia persistente e não explicada (níveis ligeira a moderadamente elevados, ~400-2000 U/L)2,7,34-37
- Níveis normais de CK não excluem a doença de Pompe34
- Miopatia dos músculos proximais inespecífica35
- Um membro da família que tem doença de Pompe3
Poderá a doença de Pompe estar dissimulada em algum dos seus doentes?
Saiba mais sobre os sinais e sintomas que impactam os doentes que vivem com a doença de Pompe.
O papel da ressonância magnética (RM) no diagnóstico
Resultados de estudos em doentes com LOPD demonstraram uma correlação entre a fraqueza muscular e achados anormais na RM e na tomografia computorizada.38
Avanços nas técnicas de imagiologia, como a RM, podem identificar atrofia e infiltração lipídica em certos músculos, bem como padrões de envolvimento muscular, apesar de não serem patognomónicos da doença de Pompe e não ajudarem especificamente a estabelecer um diagnóstico definitivo.39,40
Num estudo de história natural da doença, doentes com LOPD tinham envolvimento significativo na expansão dos pulmões durante a inspiração, em comparação com voluntários saudáveis (P=0.001)38
Reduções no volume máximo inspiratório têm um impacto direto, negativo, na função respiratória, limitando a capacidade vital forçada (CVF)41
No mesmo estudo, doentes com LOPD mostraram um declínio annual significativo na função respiratória:41
- 3,2% declínio anual na pressão inspiratória máxima (P=0.018)
- 3,8% declínio anual na pressão expiratória máxima (P<0.01)
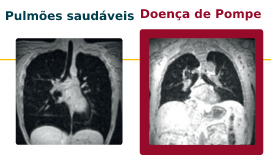
Volume inspiratório máximo (imagens compostas por RM)
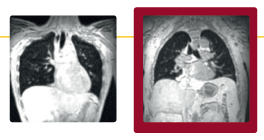
Volume expiratório máximo (imagens compostas por RM)
Imagens por RM avaliaram os movimentos máximos inspiratórios e expiratórios, enquanto os sujeitos sustiveram a respiração por 12 segundos. O volume do pulmão (retratado a preto) de cada sujeito foi estimado offline e normalizado pela razão entre o comprimento do pulmão durante a inspiração e o comprimento do pulmão durante a expiração.38
O volume inspiratório máximo pode estar consideravelmente reduzido em doentes de Pompe38
O papel da eletromiografia (EMG) no diagnóstico
Estudos demonstraram que mais de 70% dos doentes com LOPD têm um padrão miopático na EMG42
Achados da EMG sugestivos de LOPD:7
- Aumento marcado da irritabilidade da membrana muscular
- Potenciais de fibrilhação
- Ondas agudas positivas
- Descargas complexas repetitivas
- Descargas miotónicas (típicas ou atípicas)
- Breve ação da unidade motora (potencial de ação da unidade motora)
Os achados nos músculos proximais por EMG, incluindo os músculos paraespinhais, são mais propensos a revelarem anomalias
A EMG deve ser realizada nos músculos proximais e distais dos membros superiores e inferiores, bem como nos músculos torácicos paraespinhais, caso haja suspeita de LOPD.7
Diagnóstico definitivo
Jornada de diagnóstico
Apesar da jornada até ao diagnóstico da doença de Pompe ser variável, o processo geralmente envolve:2,7,34
- Avaliação clínica dos sintomas de apresentação pelo médico de família
- Referenciação a um médico especialista para investigação clínica mais aprofundada, incluindo testes laboratoriais ou clínicos adicionais
- Teste de confirmação
A análise da atividade enzimática da AGA pode identificar doentes que têm doença de Pompe34,44-46

Em doentes com IOPD, a genotipagem pode ser usada para identificar variantes genéticas comuns associadas à IOPD e ajudar na caracterização da doença. Após um diagnóstico definitivo, é recomendado o rastreio familiar.49,50
Teste da gota de sangue seco (DBS)
A doença de Pompe é confirmada por uma ausência completa ou redução marcada da atividade da AGA.51,52
A utilização de amostras de sangue, onde se incluem os DBS, é uma prática standard. As amostras sanguíneas recolhidas para testar para a doença de Pompe são pouco invasivas, precisas e geralmente permitem obter resultados em apenas alguns dias.51,52 Se a atividade enzimática da AGA estiver reduzida, o diagnóstico deve ser confirmado utilizando uma segunda amostra de sangue total e/ou através de genotipagem, de forma a confirmar a presença de dois alelos mutados no gene GAA.7,53,54
Historicamente, o teste enzimático da AGA era realizado utilizando uma cultura de fibroblastos da pele. Contudo, a colheita da amostra é relativamente invasiva e os resultados demoram cerca de 6 semanas até serem obtidos. Este longo tempo de espera não é desejado, especialmente em lactentes com uma doença que progride rapidamente.33,51,52
Apesar das biópsias musculares serem uma opção para análise da atividade da AGA, geralmente não são preferidas, uma vez que são invasivas e estão associadas a um risco elevado de falso positivos devido a um manuseamento incorreto da amostra. As biópsias musculares podem ser úteis para avaliação histológica, mas é importante notar que o conteúdo em glicogénio pode ser muito variável entre músculos, pelo que uma biópsia aparentemente normal não exclui a doença de Pompe. Desta forma, um diagnóstico de doença de Pompe deve ser sempre confirmado pela ausência ou redução da atividade da AGA e/ou por análise genética.51
Níveis patológicos da atividade enzimática da AGA são indicativos da doença de Pompe
- Em doentes com IOPD, a atividade enzimática da AGA pode estar completamente ausente; se persistir alguma atividade enzimática, é geralmente <1% do normal3
- Em doentes com LOPD, a atividade enzimática da AGA encontra-se habitualmente entre 1% - 40% do normal3
Para doentes diagnosticados com doença de Pompe, é essencial uma monitorização atempada e ativa7
Um cuidado multidisciplinar continuado pode ajudar a melhorar o prognóstico de doentes de Pompe2
Rastreio familiar
A doença de Pompe é hereditária, pelo que pode ser transmitida de pais para filhos9
Apesar da doença de Pompe ser considerada rara na população em geral, os familiares mais chegados de uma pessoa com doença de Pompe têm maior probabilidade de serem afetados pela doença ou de serem portadores.2,9 O rastreio dos familiares dos doentes com doença de Pompe pode ajudar ao diagnóstico atempado e a uma monitorização mais eficaz da doença.2,4
Os irmãos de um caso índice não são os únicos em risco de ser afetados pela doença de Pompe
Saiba porquê neste vídeo
.png/jcr:content/thumb-video-rastreio-familia%20(1).png)
Vídeo McIntosh et al 20202
Jornada do doente – irmãos Sean e Cheryl
O Sean e a Cheryl são irmãos e partilham a sua experiência com a doença de Pompe. Após o Sean saber qual o seu diagnóstico, encorajou a irmã a ser testada e ela também foi diagnosticada com doença de Pompe. Juntos partilham a sua perspetiva quanto à importância de se apoiarem mutuamente e do rastreio familiar.
.png/jcr:content/thumb-video-cheryl%20(1).png)
Living with Pompe disease Sean's and Cheryl's story
Referências
- Kishnani PS, et al; on behalf of the Pompe Registry Boards of Advisors.Am J Med Genet. 2013;161A(10):2431-2443.
- Kishnani PS, et al; ACMG Work Group on Management of Pompe Disease. Genet Med. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3.
- Kishnani PS, et al../ Pediatr. 2004;144(5 suppl):S35-S43.
- Rigter T, et al. Mol Genet Metab. 2012;107(3):448-455. doi:0.1016/1.ymgme.2012.09.017.
- Schuller A, et al. Am J Med Genet C Semin Med Genet. 2012;160C(1.):80-88. doi:10.1002/ajmg.c.31322.
- van Capelle CI, et al. Orphanet J Rare Dis. 2016;11(1):65. doi:10.1186/s13023-016-0442-y.
- American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160.doi:10.1002/mus.21393.
- Kishnani PS, et al; Infantile-Onset Pompe Disease Natural History Study Group. J Pediatr. 2006;148(5):671-676. doi:10.1016/j.jpeds.2005.11.033.
- Hirschhorn R, et al. In: Scriver CR, et al, eds. The Metabolic Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420.
- Fusco AF, et al. Int J Mol Sci. 2020;21(6):2256. doi:10.3390/ijms21062256.
- Hereditary myopathy with early respiratory failure. Genetics Home Reference website. https://ghr.nlm.nih.gov/condition/hereditary-myopathy-with-early-respiratory-failure. Reviewed September 2018. Accessed December 15, 2020.
- Barohn RJ, et al. Neurol Clin. 2014;32(3):569-vii. doi:10.1016/j.nc1.2014.04.008.
- Chaudhuri A, et al. Lancet 2004;363(9413):978-988. doi:10.1016/50140- 6736(04)15794-2.
- Jaradeh S. Muscle disorders affecting oral and pharyngeal swallowing. GI Motility Online website. https://vvww.nature.com/gimo/contents/pWfull/gimo35.html. Published May 16, 2006. Accessed December 15, 2020.
- Gilchrist 1M. Semin Respir Crit Cam Med. 2002;23(3):191-200. doi:10.1055/s-2002-33027.
- Ozawa E, et al. Mol Cell Biochem. 1999;190:143-151.
- Mah JK, et al. Neuromuscul Disord. 2014;24(6):482-491. doi:10.1016/j.nmd.2014.03.008.
- Barnabei MS, et al. Compr Physiol. 2011;1(3):1353-1363. doi:10.1002/cphy.c100062.
- Limb-girdle muscular dystrophy. Genetics Home Reference website. https://ghtnlm.nih.gov/condition/limb-gird le-muscular-dystrophy. Reviewed September 2019. Accessed December 15, 2020.
- Pegoraro E, et al. In: Pagan RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1408/. Published June 8,2000. Updated August 30, 2012. Accessed December 15, 2020.
- Siciliano G, et al. Acta Myol. 2015;34(1):3-8.
- Myasthenia gravis. Genetics Home Reference website. https://ghr.nlm.nih.gov/condition/myastheniagravis. Published February 27, 2017. Accessed December 15, 2020.
- Myasthenia gravis: what is it? Harvard Health Publishing website. https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Published December 2018. Accessed December 15, 2020.
- Smoyer-Tomic KE, et al. BMC Musculoskeletal Disord. 2012;13:103. doi:10.1186/1471- 2474-13-103.
- Inflammatory myopathies information page. National Institute of Neurological Disorders and Stroke website. https://www.ninds.nih.gov/Disorders/All-Disorders/1 nflammatory-Myopathies-I nformation-Page. Published February 27, 2017. Accessed December 15, 2020.
- Gazeley DJ, et al. Ther Adv Musculoskeletal Disord. 2011;3(6):315-324. doi:10.1177/1759720X11415306.
- Polymyositis. Johns Hopkins Medicine website. https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Published February 27, 2017. Accessed December 15,2020.
- Mastaglia FL, et al. Rheum Dis Clin North Am. 2002;28(4):723-741. doi:10.1016/s0889-857x(02)00021-2.
- Rudnik-Schoneborn 5, et al. Eur NeuroL 1998;39(3):154-162. doi:10.1159/000007926.
- Davis RH, et al../ Child Neural. 2014;29(11):1467-1472. doi:10.1177/0883073813503988.
- D'Souza RS, et al. Circ Heart Fail. 2014;7(5):843-849. doi:10.1161/CIRCHEARTFAILURE.114.001105.
- Fu L, et al. Korean Circ J. 2013;43(12):785-792. doi:10.4070/kcj.2013.43.12.785.
- Leslie N, et al. GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK1261/. Published August 31, 2007. Updated May 11, 2017. Accessed December 15, 2020.
- Toscano A, et al. Acta Myol. 2013;32(2):78-81.
- Preisler N, et al. Mol Genet Metab. 2013;110(3):287-289. doi:10.1016/j.ymgme.2013.08.005.
- Manganelli F, et al.Acta Myol. 2013;32(2):82-84.
- Moghadam-Kia 5, et al. Cleve Clin J Med. 2016;83(1):37-42. doi:10.3949/ccjm.83a.14120.
- Wens SCA, et al. BMC Pulmonary Medicine. 2015;15:54. doi:10.1186/512890-015-0058-3.
- Figueroa-Bonaparte 5, et al. PLoS One. 2016;11(10):e0163493. doi:10.1371/journal.pone.0163493.
- Paoletti M, et al. Front Neural. 2019;10:78. doi:10.3389/fneur.2019.00078.
- van der Beek NAME, et al. Mol Genet Metab. 2011;104:129-136.
- Killer-Felber W, et al. Neuromuscul Disord. 2007;17(9-10):698-706. doi:10.1016/j.nmd.2007.06.002.
- Wagner M, et al. Neuromuscul Disord. 2013;23(1):89-92. doi:10.1016/j.nmd.2012.09.004.
- Behjati 5, et al. Arch Dis Child Educ Pratt Ed. 2013;98(6):236-238. doi:10.1136/archdischild-2013-304340.
- Okumiya T, et al. Mol Genet Metab. 2006;88(1):22-28. doi:10.1016/j.ymgme.2005.10.016.
- Cupler EJ, et al; AANEM Consensus Committee on Late-Onset Pompe Disease. Muscle Nerve. 2012;45(3):319-333. doi:10.1002/mus.22329.
- Bodamer OA, et al; on behalf of the Pompe Disease Newborn Screening Working Group. Pediatrics. 2017;140(suppl 1):S4-S13. doi:10.1542/peds.2016-0280C.
- Recommended uniform screening panel. Health Resources and Services Administration website. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html. Reviewed February 2020. Accessed December 15, 2020.
- Taverna 5, et al. Aging (Albany NY). 2020;12(15):15856-15874. doi:10.18632/aging.103794.
- Atherton AM, et al. Pediatrics. 2017;140(suppl 1):546-550. doi: 10.1542/peds.2016-0280F.
- Winchester B, et al. Mol Genet Metab.2008;93(3):275-281. doi: 10.1016/j.ymgme.2007.09.006.
- Zhang H, et al. Genet Med.2006;8(5):302-306. doi: 10.1097/01.gim.0000217781.66786.9b.
- van der Hoeg AT, et al. Eur J Neurol. 2017;24(6):768-e31. doi:10.1111/ene.13285.
- Llerena JC Jr, et al.Arq Neuropsiquiatr. 2016;74(2):166-176. doi:10.1590/0004-282X20150194












