Os danos iniciam-se muito antes do surgimento dos primeiros sintomas
Diagnostique e monitorize atempadamente a doença de Pompe, permitindo aos seus doentes estarem um passo à frente da doença

A doença de Pompe, também conhecida como glicogenose tipo II, é uma doença neuromuscular genética e progressiva que pode afetar doentes de todas as idades1-3
A doença de Pompe pode causar danos musculares que podem resultar em falência respiratória, perda da função motora, falência cardíaca e morte prematura.1,2,4,5
A doença de Pompe é heterogénea, podendo variar na sintomatologia, na idade de início dos sintomas ou na taxa de progressão.1,6
Os doentes com doença de Pompe apresentam frequentemente sinais de envolvimento das funções motora e respiratória1,4,5
IDoença de Pompe de forma infantil (IOPD, Infantile-onset Pompe disease)
- Prevalência: 1 em 138.000 pessoas7
- Apresentação: durante o primeiro ano de vida1,8
- Progressão: de progressão rápida e, se não tratada, resulta em morte até aos 2 anos de idade, devido mais frequentemente, a falência respiratória2
Doença de Pompe de Início Tardio (LOPD, Late-onset Pompe disease)
- Prevalência: 1 em 57.000 pessoas7
- Apresentação: a qualquer altura, durante a infância ou idade adulta1
- Progressão: de progressão constante, podendo resultar em morte premature, devido mais frequentemente, a falência respiratória1
Aprenda a reconhecer os sinais e sintomas da doença de Pompe
Aproximadamente 1 em 40.000 pessoas a nível mundial têm a doença de Pompe7
Manifestações Comuns
IOPD
- Doentes com IOPD podem apresentar cardiomiopatia, hipotonia, fraqueza muscular e dificuldades respiratórias, alimentares e de atingimento dos marcos de desenvolvimento.1,8
LOPD
- 77% dos doentes com LOPD apresentam fraqueza dos músculos proximais e intolerância ao exercício físico4
LOPD
- 55% dos doentes com LOPD apresentam insuficiência respiratória e fraqueza dos músculos proximais4
Em doentes com LOPD, a causa mais comum de morte é a falência respiratória9
O declínio contínuo da função respiratória pode ocorrer sem tratamento, pelo que é necessária a monitorização da doença de forma atempada e ativa.1,6,9-11
Descoberta e genética
Na doença de Pompe, a deficiência da atividade enzimática da α-glucosidase ácida (AGA) culmina em danos musculares, progressivos e debilitantes1-3
Descoberta
A doença de Pompe foi descrita pela primeira vez em 1932 pelo patologista Holandês Johannes C. Pompe, que relatou o caso de uma criança de 7 meses de idade que morreu de hipertrofia cardíaca idiopática. Verificou-se que esta criança tinha uma acumulação massiva de glicogénio em muitos tecidos, mas predominantemente nos músculos esquelético e cardíaco. Apesar da doença da doença de Pompe ter sido diagnosticada pela primeira vez num lactente, esta pode afetar tanto doentes pediátricos como adultos.12,13
Deficiência
Em 1963, a doença de Pompe foi associada a uma deficiência hereditária da enzima AGA, presente no lisossoma e responsável pelo catabolismo de glicogénio em glucose.12 A doença de Pompe é uma doença autossómica recessiva.1 Variantes patogénicas hereditárias no gene que codifica a enzima AGA - gene GAA 17q25.2-q25.3, no cromossoma 17 – causam a deficiência marcada ou a ausência de atividade enzimática em doentes de Pompe.12,14
Destruição
Na doença de Pompe, a acumulação contínua de glicogénio, devido ao défice de atividade enzimática da AGA, causa um ingurgitamento e rutura dos lisossomas, resultando em danos celulares. Por sua vez, estes danos levam a uma degeneração progressiva dos músculos esqueléticos e respiratórios (em conjunto com o músculo cardíaco, principalmente em lactentes), resultando eventualmente em perda de função.12,15
Acumulação de glicogénio em células de um doente de Pompe

A. Miofibrilhas
B. Glicogénio lisossomal entre miofibrilhas
C. Acumulação de glicogénio lisossomal na periferia da célula
D. Lisossomas com rutura da membrana e formação de “lagos” de glicogénio
Em doentes com doença de Pompe, a acumulação de glicogénio e consequente patologia muscular ocorrem habitualmente antes da deteção de sinais ou sintomas.1,3,16,17
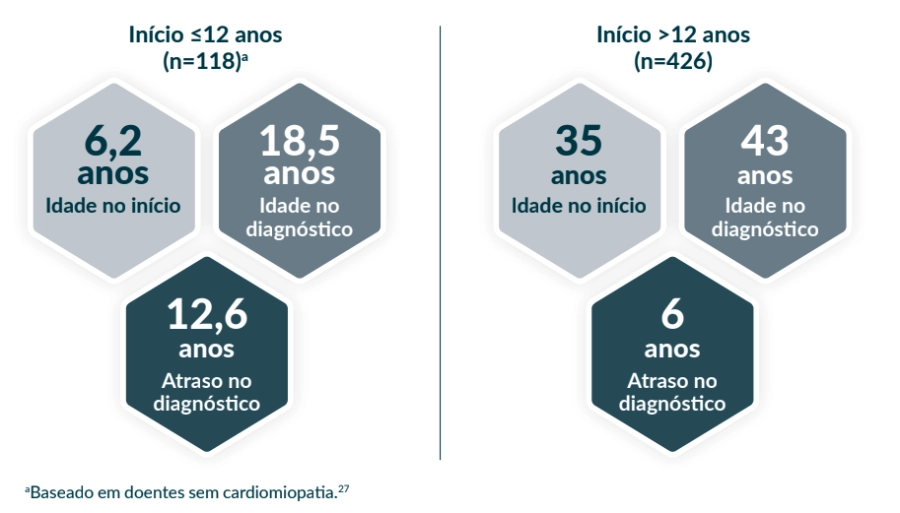
Atraso no diagnóstico
Doentes com LOPD podem experienciar um atraso de 6 a 12,6 anos até receberem um diagnóstico definitivo18
Sem uma monitorização apropriada da doença, a LOPD pode resultar em danos musculares progressivos e potencialmente irreversíveis, em declínio da função respiratória e em mobilidade reduzida1,2,19,20
Intervalo de diagnóstico mediano em doentes com LOPD18,a
Não perca tempo
O diagnóstico e tratamento atempados são essenciais para gerir os sintomas e para melhorar o prognóstico dos doentes de Pompe3,21

Diagnostique e monitorize atempadamente a doença de Pompe, ajudando a reduzir a carga para o doente1-3
A acumulação progressiva de glicogénio e os danos musculares contínuos necessitam de um diagnóstico atempado e da monitorização ativa dos doentes com doença de Pompe.19
Referências
- Kishnani PS, et al; ACMG Work Group on Management of Pompe Disease. Genet Med. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3.
- Kishnani PS, et al. J Pediatr. 2004;144(5 suppl):S35-S43.
- American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160. doi:10.1002/mus.21393.
- Schüller A, et al. Am J Med Genet C Semin Med Genet. 2012;160C(1):80-88. doi:10.1002/ajmg.c.31322.
- van Capelle Cl, et al. Orphanet J Rare Dis. 2016;11(1):65. doi:10.1186/s13023-016-0442-y.
- Chien YH, et al. J Pediatr. 2011;158(6):1024-1027.e1. doi:10.1016/j.jpeds.2010.11.053.
- Ausems MGEM, et al. Eur J Hum Genet. 1999;7(6):713-716. doi:10.1038/sj.ejhg.5200367.
- Kishnani PS, et al; Infantile-Onset Pompe Disease Natural History Study Group. J Pediatr. 2006;148(5):671-676. doi:10.1016/j.jpeds.2005.11.033.
- Winkel LPF, et al. J Neurol. 2005;252(80):875-884. doi:10.1007/s00415-005-0922-9.
- Schoser B, et al. J Neurol. 2017;264(4):621-630. doi:10.1007/s00415-016-8219-8.
- Alejaldre A, et al. Neuromuscul Disord. 2012;22(suppl 2):S148-S154. doi:10.1016/j.nmd.2012.05.011.
- Hirschhorn R, et al. In: Scriver CR, et al, eds. The Metabolic Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420.
- Fukuda T, et al. Muscle Nerve. 2007;7:71-77.
- Pittis MG, et al. Acta Myologica. 2007;26(1):67-71.
- Müller-Felber W, et al. Neuromuscul Disord. 2007;17(9-10):698-706. doi:10.1016/j.nmd.2007.06.002.
- Thurberg BL, et al. Lab Invest. 2006;86(12):1208-1220.
- Di Rocco M, et al Acta Myol. 2007;26(1):42-44.
- Kishnani PS, et al; on behalf of the Pompe Registry Boards of Advisors. Am J Med Genet. 2013;161A(10):2431-2443.
- Rigter T, et al. Mol Genet Metab. 2012;107(3):448-455. doi:0.1016/j.ymgme.2012.09.017.
- Hagemans ML, et al. Neurology. 2005;64(12):2139-2141.
- Al Jasmi F, et al; the MENA Pompe Working Group. BMC Neurology. 2015;15:205. doi:10.1186/s12883-015-0412-3.











