
Fabry
Descubriendo la enfermedad de Fabry
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosomal ligado al cromosoma X que afecta a hombres, mujeres y niños de todas las etnias.1,2
¿Qué es la enfermedad de Fabry?

Generalidades de las Enfermedad de Fabry
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosomal ligado al cromosoma X que afecta a hombres, mujeres y niños de todas las etnias.1,2 Su prevalencia se estima entre 1: 40.000.1,3
Es un trastorno multisistémico progresivo que puede comprometer órganos vitales como los riñones, el corazón y el cerebro incrementando la morbimortalidad de los pacientes.2,4
Los primeros signos y síntomas de la enfermedad de Fabry pueden comenzar en la infancia o la adolescencia incluyendo dolor, trastornos gastrointestinales, angioqueratomas, hipohidrosis y proteinuria, entre otros.5 A medida que progresa puede producir deterioro en la función renal, miocardiopatía hipertrófica y aumento del riesgo a desarrollar eventos graves tales como enfermedad renal terminal, accidente cerebrovascular, insuficiencia cardíaca, arritmias y muerte prematura.1,5
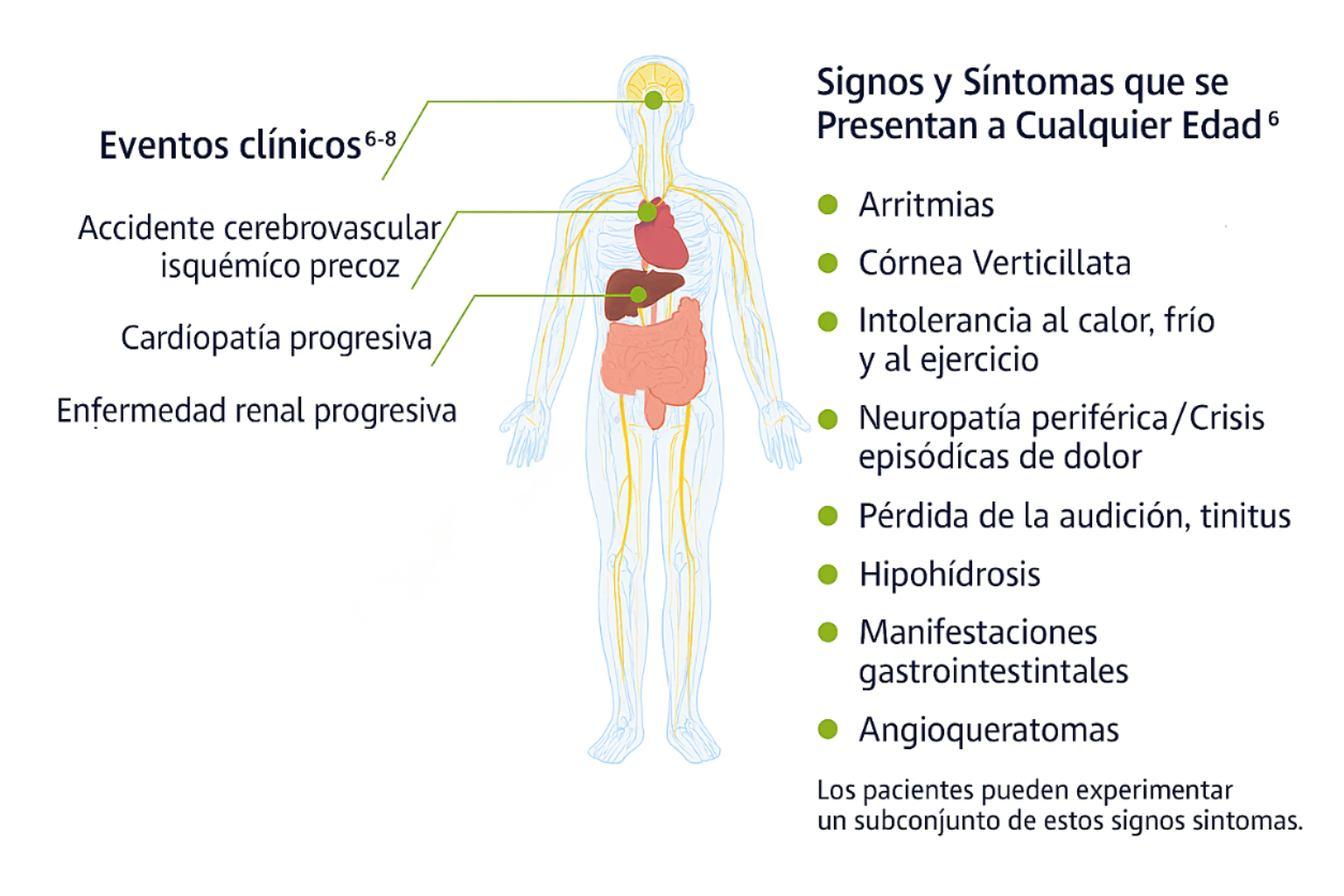
La enfermedad de Fabry es multisistémica. Conoce más acerca de sus manifestaciones:
Signos y síntomas por etapa de la vida
Los pacientes pueden experimentar algunos o todos estos síntomas.

Fisiopatología
En la enfermedad de Fabry, las mutaciones en el gen GLA, ubicado en el cromosoma X, producen defectos en la síntesis y/o función de la alfa-galactosidasa A (α- Gal A).9
La α- Gal A es una enzima lisosomal que metaboliza la globotriaosilceramida (también conocida como GL-3 o Gb3) y evita que se acumule.10
La deficiencia total o parcial de α- Gal A conduce a la acumulación progresiva de glicoesfingolípidos, particularmente GL-3, en los lisosomas de numerosos tipos celulares.5
En la enfermedad de Fabry, la acumulación de GL-3 comienza en el útero, y continúa durante toda la vida.1,2 Con el avance de la edad su acumulación, particularmente en el endotelio vascular, conduce a insuficiencia renal, enfermedad cardíaca, accidente cerebrovascular y muerte prematura, generalmente en la cuarta o quinta década de la vida.4,5,10,11
Las manifestaciones clínicas de la enfermedad de Fabry abarcan un amplio espectro en términos de severidad y generalmente están correlacionadas con el nivel de actividad residual de α- Gal A
Generalidades genéticas
La enfermedad de Fabry es un trastorno genético; en consecuencia la detección familiar es de vital importancia.
Por cada paciente diagnosticado con Fabry, hay una familia que está en riesgo
Se han identificado más de 900 mutaciones en el gen GLA asociado con la enfermedad de Fabry.9,12 La mayoría de las familias tienen mutaciones específicas.2
Por cada paciente índice diagnosticado, se puede identificar a un promedio de 5 miembros adicionales de la familia que están afectados.
Como es una enfermedad ligada al cromosoma X, los hombres y las mujeres pueden transmitir la mutación genética que causa la enfermedad de Fabry. Además es importante destacar que las mujeres no son simplemente portadoras.1,5,13 Los hombres tienen una probabilidad del 100% de transmitir el gen mutado a sus hijas y no a sus hijos varones, mientras que las mujeres con enfermedad de Fabry tienen una probabilidad del 50% de transmitir el gen mutado a cada hija e hijo.5
Las mujeres con el gen GLA mutado pueden desarrollar Fabry, pero las manifestaciones pueden ser heterogéneas debido a la inactivación aleatoria del cromosoma X específica de órganos y tejidos.
Fenotipos14-16
Se han descripto dos formas de presentación para la enfermedad de Fabry: el fenotipo clásico de afectación multisistémica (Enfermedad de Fabry tipo 1) y las formas de inicio tardío (Enfermedad de Fabry tipo 2) que suelen afectar un solo órgano blanco.

El camino al diagnóstico
En un estudio basado en el Registro de Fabry, los retrasos en el diagnóstico y el diagnóstico erróneo fueron comunes debido a la naturaleza inespecífica y heterogénea de los primeros síntomas de la enfermedad de Fabry.8
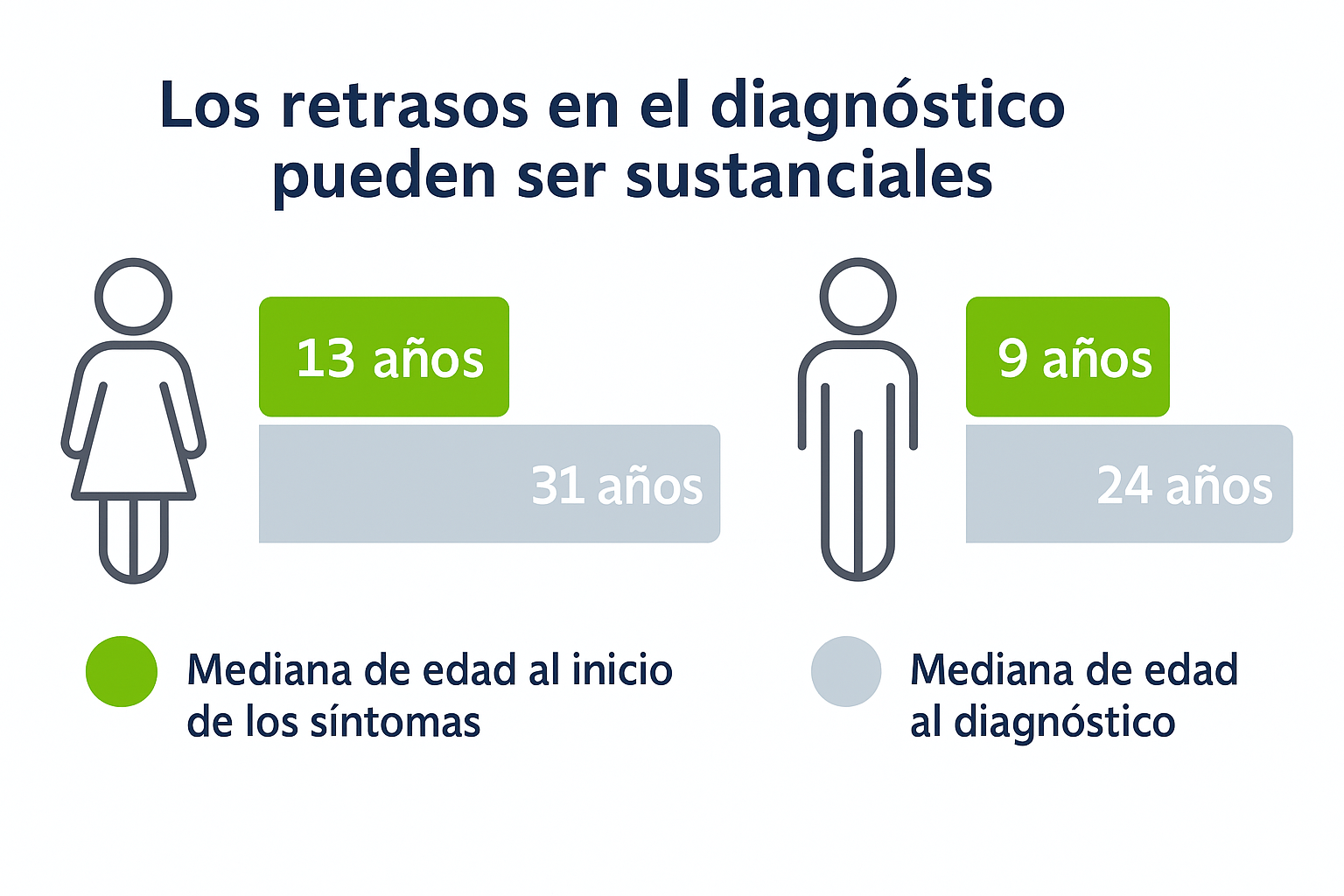
A menudo, la enfermedad de Fabry se diagnostica erróneamente y se confunde con artritis reumatoidea o artritis idiopática juvenil, fiebre reumática, síndrome de Raynaud, lupus eritematoso sistémico o esclerosis múltiple.2,5
Conectar los síntomas aparentemente no relacionados con la enfermedad de Fabry puede ayudar a evitar los retrasos diagnósticos e iniciar la terapia a tiempo.10
La importancia de la detección de la enfermedad de Fabry en las poblaciones de alto riesgo
Aunque a la enfermedad de Fabry se la considera una enfermedad poco frecuente, la prevalencia en los pacientes con ciertos trastornos como enfermedad renal crónica inexplicada, miocardiopatía hipertrófica, y accidente cerebrovascular prematuro, es mayor que en la población general.17-19 Por consiguiente, es importante considerar la EF dentro de los diagnósticos diferenciales.
Las mujeres con enfermedad de Fabry
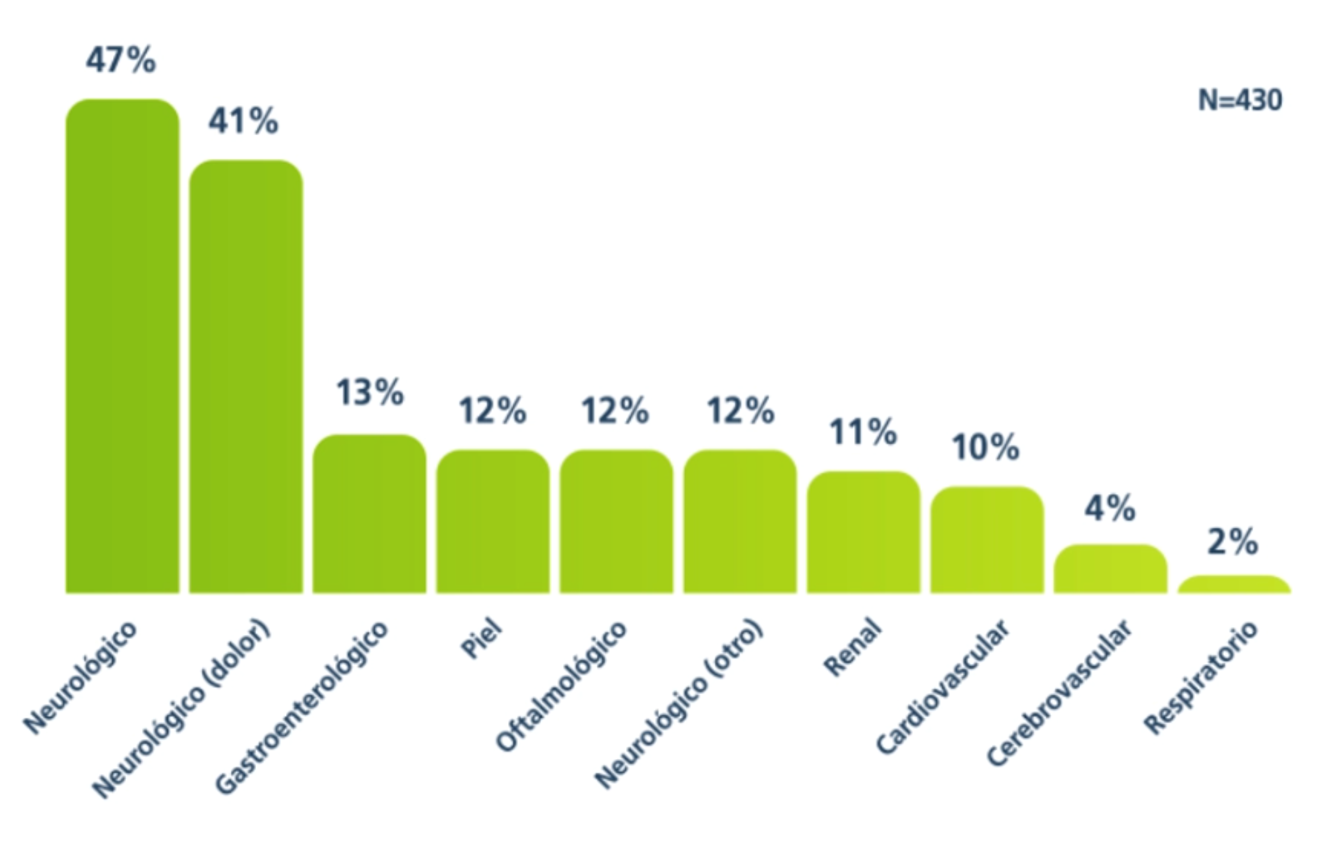
Según el Registro, el 69,4% de las mujeres con la enfermedad de Fabry desarrollan síntomas relacionados con la afección con el tiempo.8
Se sabe que las mujeres con EF presentan un curso clínico heterogéneo, por lo que generalmente sus síntomas se desarrollan en formas más tardía a diferencia de los hombres.13 Este rango de heterogeneidad en la sintomatología hace que el diagnóstico de Fabry en mujeres sea especialmente desafiante.
En las mujeres, esta variabilidad en la presentación de la EF se debe a los patrones de inactivación aleatorios del cromosoma X. Cada órgano en el cuerpo de una mujer tiene su propio patrón de inactivación aleatoria y conduce a variaciones en la expresión de la enfermedad.13
La actividad enzimática en las mujeres puede estar levemente disminuida o en rangos normales. En un estudio transversal de 57 mujeres heterocigotas sintomáticas, algunas con actividad enzimática normal presentaban anomalías cardíacas, renales o cerebrovasculares.22
Compromiso por sistema orgánico en las mujeres con la enfermedad de Fabry4
Referencias
- Desnick RJ, et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2014:1-64.
- Desnick RJ. Ann Intern Med. 2003;138(4):338-346.
- Laney DA, et al. J Genet Couns. 2008;17(1):79-83
- Eng CM, et al. J Inherit Metab Dis. 2007;30(2):184-192.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49.
- Barba-Romero MA, et al. Int J Clin Pract. 2011;65(8):903-910.
- Mehta A. QJM. 2002;95(10):647-653.
- Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112-128.
- Nagueh SF. Circulation. 2014;130(13):1081-1090.
- Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-1607.
- Sims K. et al. Stroke. 2009:40(3):788-794.
- The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed January 19, 2018.
- Wang RY et al. Genet Med 2007;9(1):34-45.
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-427.
- Germain D. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
- Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, Elliott PM, Linthorst GE, Wijburg FA, Biegstraaten M, Hollak CE. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol. 2017 May;28(5):1631-1641.
- Nakao S, et al. Kidney Int. 2003;64(3):801.807.
- Linthorst GE, et al. Nephrol Dial Transplant. 2003;18(8):1581-1584.
- Kotanako P, et al. J Am Soc Nephrol. 2004;15(5):1323-1329.
- Gupta S, et al. Medicine (Baltimore). 2005;84(5):261-268.
- Shelley ED, et al. Pediatr Dermatol 1995;12(3):215-219.
- Fabry Registry Annual Report 2010. Available at: www.fabry.org/fsig.nsf/PDFs/PDFsR/$File/2010_Annual_Report.pdf. Accessed January 22. 2018.
- Ramaswami U, et al. Clin J Am Soc Nephrol. 2010;5(2):365-370.
- Ichinose M, et al. Clin Exp Nephrol. 2005;9(3):228-232
- Bekri S, et al. Nephrol Clin Pract 2005:101(1):c33-38.
- Coresh J, et al. JAMA. 2007;298(3):2038-2047.
- Waldek S, et al. BMC Nephrol. 2014:15(72):1-15.
- Najafian B, et al. Kidney Int. 2011;79(6):663.670.
- Torra R. Kidney Int. Suppl. 2008;(Suppl. 111):S29-S32.
- Schiffmann R, et al. Nephrol Dial Transplant. 2009.24(7) 2102-2111.
- Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505.517.
- Linthorst GE, et al. J Med Genet. 2010;47(4):217-222.
- Monserrat L, et al. Coll Cardiol. 2007;50(25):2399-2403.
- van der Tol L, et al. J Med Genet. 2014;51(1):1-9.
- Yousef Z, et al. Eur Heart J. 2013;34(11):802-808.
- Eng CM, et al. Genet Med. 2006;8(9):539-548.
- Linhart A, et al. Eur Heart J. 2007;28(10):1228-1235.
- Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-1099.
- Kampmann C, et al. Int J Cardiol. 2008:130(3):367-373.
- Weidemann F, et al. Orphanet J Rare Dis. 2013:8(116).
- Elliott PM, et al. Eur Heart J. 2014;35(39):2733-2779.
- Fellgiebel A, et al. Lancet Neurol. 2006;5(9):791-795.
- Hilz MJ. Clin Ther. 2010;32 (suppl C):S93.
- Bottcher T. et al. Plos ONE. 2013:8(8):e71894.
- Cable WJL, et al. Neurology. 1982;32(5):498-502.
- Uceyler N, et al. Clin J Pain. 2014;30(10):915-920.
- Fellgiebel A, et al. Neurology. 2009;72(1):63-68.
- Moore DF, et al. Brain Res Bull. 2003;62(3):231-240.
- Cole AL, et al. J Inherit Metab Dis. 2007:30(6):943-951.
- Samiy N, et al. Surv Ophthalmol. 2008;53(4): 416-423.
- Hopkin RJ, et al. Pediatr Res. 2008;64(5):550-555.
- MacDermot KD, et al. J Med Genet. 2001a;38(11):750-760.
- MacDermot KD, et al. J Med Genet. 2001b;38(11):769-775.
- Manger B, et al. Clin Rheumatol. 2007;26(3):335-341