

%20new%20(1).2023-10-09-16-08-30.jpg)

Patrón hereditario de la enfermedad de Fabry
En la enfermedad de Fabry, las madres afectadas tienen un riesgo del 50% de transmitir el gen GLA mutado a sus hijos independientemente del género, mientras que los padres afectados transmiten el gen GLA defectuoso a todas sus hijas y a ninguno de sus hijos.1
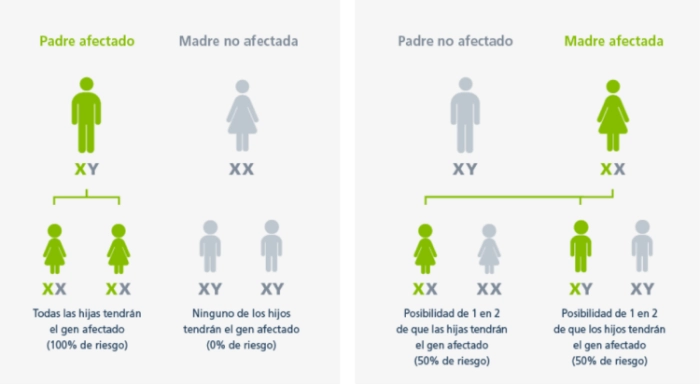
En la enfermedad de Fabry, las madres afectadas tienen un riesgo del 50% de transmitir el gen GLA mutado a sus hijos independientemente del género, mientras que los padres afectados transmiten el gen GLA defectuoso a todas sus hijas y a ninguno de sus hijos.1
La importancia del árbol Genealógico
Una vez que se diagnostica un caso índice de la enfermedad de Fabry, esto posibilita que los familiares pre-sintomáticos puedan ser diagnosticados a una edad más joven, lo que permite un manejo más temprano de la enfermedad. La detección familiar y el análisis de los antecedentes familiares pueden ser especialmente útiles para identificar a las mujeres en riesgo de contraer la enfermedad de Fabry.
La evaluación familiar puede ayudar a encontrar los pacientes en una etapa más temprana en el curso de su enfermedad.

El diagnóstico de cualquier paciente con Fabry debe ser seguido por un estudio genealógico completo en forma de análisis del árbol familiar y la realización de exámenes a los otros miembros de la familia que están en riesgo de la enfermedad de Fabry. Un árbol familiar es el primer paso para identificar a otros miembros de la familia con Fabry.
¿Necesitas asesoramiento?
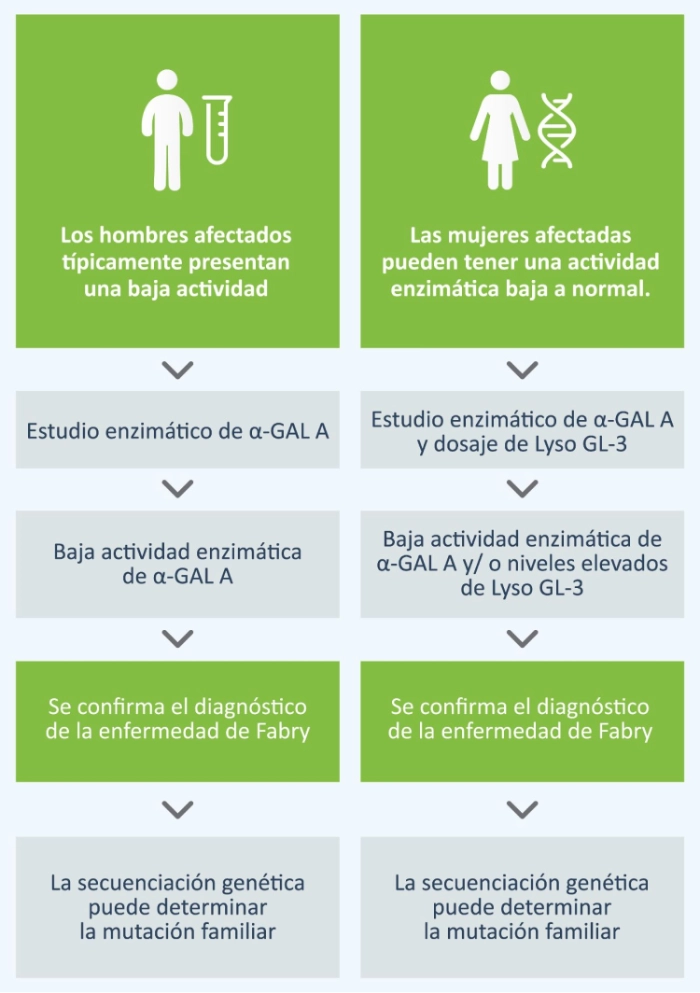
Técnicas diagnósticas
En los hombres, el diagnóstico se realiza mediante la prueba de actividad enzimática de α- galactosidasa A (α- GAL) en plasma o sangre seca. Debido a que las mujeres con enfermedad de Fabry pueden tener actividad en rango bajo a normal se recomienda también realizar el dosaje del biomarcador específico Lyso Gl-3 en gota de sangre seca.
La identificación de una mutación patogénica mediante el estudio del gen GLA es relevante para confirmar el diagnóstico en mujeres y hombres.1 Los resultados pueden ser útiles para comprender las correlaciones entre el genotipo y el fenotipo.
Mutaciones más frecuentes en Argentina
La enfermedad de Fabry se produce a partir de la presencia de una variante patogénica en el gen GLA. Se han identificado más de 900 mutaciones hasta el momento.
Las distintas variantes o mutaciones que existen producen distintos fenotipos: en general los pacientes con enfermedad de Fabry con una actividad enzimática de α-GAL A nula o casi nula (<1%) muestran síntomas severos (fenotipo clásico), mientras que los pacientes con actividad enzimática residual muestran síntomas de aparición más tardía con afectación cardiológica o renal (fenotipo tardío).
Se ha descripto mucha variabilidad fenotípica incluso dentro de una misma familia, sugiriendo que además del genotipo otros factores podrían estar involucrados en la clínica de la enfermedad. Además de los cambios genotípicos (variantes en el gen GLA), se deben considerar los cambios epigenéticos para explicar las diversidades fenotípicas entre los pacientes con las mismas mutaciones.4
Por ende, la enfermedad de Fabry no solo es heterogenética (variabilidad de mutaciones) sino también heterofenotípica. 5 Las 10 mutaciones más prevalentes hoy por hoy en Argentina son las siguientes. Algunas de ellas solo afectan a una sola familia y otras a varias familias en el país y la mayoría producen un fenotipo clásico.
.2023-10-09-14-08-01.webp)
- Barriales-Villa, Gimeno-Blanes, Zorio-Grima, Ripoll-Vera, Evangelista-Masip, Moya-Mitjans, Serratosa-Fernández, Albert-Brotons, García-Pinilla, García-Pavía. Plan of Action for Inherited Cardiovascular Diseases: Synthesis of Recommendations and Action Algorithms. Rev Esp Cardiol 2016;69(3):300-9. doi: 10.1016/j.rec.2015.11.029.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006 Apr 11;113(14):1807-16. DOI: 10.1161/CIRCULATIONAHA.106.174287.
- Perry Elliott, Bert Andersson, Eloisa Arbustini, Zofia Bilinska, Franco Cecchi, Philippe Charron, Olivier Dubourg, Uwe Kühl, Bernhard Maisch, William J. McKenna, Lorenzo Monserrat, Sabine Pankuweit, Claudio Rapezzi, Petar Seferovic, Luigi Tavazzi, Andre Keren. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008;29:270-6. doi.org/10.1093/eurheartj/ehm342 .
- Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 2013 Dec 3;62(22):2046-72. doi: 10.1016/j.jacc.2013.08.1644. Epub 2013 Nov 18. No abstract available. Erratum in: J Am Coll Cardiol 2014 Jan 21;63(2):191-4.
- Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L, Tendera M, Maupain C, et al. EORP Cardiomyopathy Registry Investigators. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J 2018;39(20):1784-1793. doi: 10.1093/eurheartj/ehx819.
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/America
Un enfoque holístico para la atención de la enfermedad de Fabry
Debido a que la enfermedad de Fabry se caracteriza por ser crónica y progresiva, con afectación de múltiples órganos blancos, la evaluación del paciente debe ser realizada por varias especialidades como: nefrólogos, neurólogos, cardiólogos, genetistas y otros profesionales de la salud.1
Inicio de tratamiento
El inicio del tratamiento (según consensos nacionales e internacionales) dependerá de la forma de presentación de la enfermedad, es decir que no se utilizan los mismos criterios en las formas clasicas y tardias.
En pacientes pediátricos con fenotipo clásico y presencia de signos y síntomas característicos de la enfermedad de Fabry, se recomienda iniciar terapia de reemplazo enzimático sin distinción de edad ni género. En pacientes pediátricos varones con fenotipo clásico asintomáticos se recomienda iniciar terapia de reemplazo enzimático entre los 8-10 años de edad, en base a los resultados de biopsias renales reportados en la literatura científica, donde todos los pacientes estudiados mostraron daño podocitario temprano y en muchos casos irreversible (fibrosis).2-3-4
En pacientes pediátricos varones o niñas con variantes del adulto la terapia de reemplazo enzimático puede diferirse hasta que se presenten signos o síntomas característicos tempranos de la enfermedad o se demuestre daño tisular activo en la biopsia renal y/o cardíaca. Esto suele ser en la gran mayoría de los casos luego de llegar a la edad adulta.2-3
Recomendaciones para el inicio de tratamiento en pacientes adultos según recomendaciones de las guías argentinas.2

Seguimiento del paciente Fabry
Si bien los estudios, análisis y escalas para el seguimiento en la enfermedad han sido publicados reiteradamente2, no todos son necesarios para evaluar la respuesta al tratamiento en la práctica clínica diaria y quedarán a criterio del médico tratante.
Actualmente, el Consenso Europeo para la Evaluación de Metas Terapéuticas concluye que siendo la enfermedad de Fabry una enfermedad progresiva, las metas terapéuticas serán diferentes en relación con el momento de inicio del tratamiento, siendo que en pacientes asintomáticos u oligosintomáticos un inicio temprano de la terapia puede prevenir el desarrollo de síntomas, o aún revertirlos. En pacientes con daño orgánico moderado, el objetivo terapéutico será principalmente estabilizar la progresión natural de la enfermedad. Por último, en pacientes con daño orgánico severo el tratamiento busca estabilizar el compromiso o enlentecer la aparición de complicaciones graves.5
Las herramientas para el seguimiento pueden ser clasificadas en:
- Valoración clínica
- Cuantificación de la reducción del biomarcador
- Seguimiento de la funcionalidad de cada órgano
En la práctica pediátrica, donde el daño de los órganos nobles (cerebro, corazón y riñón) no suelen estar presentes o son mínimos, se considera como herramientas útiles a las escalas de cuantificación del dolor neuropático (escala visual análoga, inventario de dolor resumido, etc.), escala de cuantificación de daño gastrointestinal (similares a las utilizadas en intestino irritable) y escalas de calidad de vida.
Actualmente se considera que el biomarcador Lyso GL-3 no solo es una herramienta diagnóstica, sino una comorbilidad en sí dada su toxicidad y una herramienta para monitorear la respuesta al tratamiento. El consenso Europeo de metas terapéuticas concluye: “el Lyso GL-3 plasmático debe ser reducido a los menores valores posibles durante los primeros 6 meses de tratamiento. La falla en lograr esta meta puede ser una indicación de intensificar la terapia de reemplazo enzimático”.5
La funcionalidad de cada órgano ha sido publicada en las guías nacionales e internacionales, donde no sólo se describe el estudio, sino la frecuencia en que deben ser solicitados en relación con el estado funcional de cada órgano y sistema.6
Estudios de seguimiento²
%201.2023-10-09-14-23-24.svg)
Pacientes diagnosticados no tratados
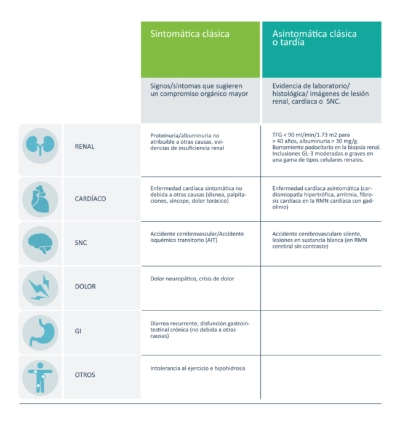
Mujeres
Las mujeres asintomáticas deben ser monitoreadas para detectar el desarrollo temprano de síntomas.6
Un porcentaje considerable de mujeres puede experimentar complicaciones
mayores y deben ser monitoreadas.7
Ya se ha observado la afección de órganos vitales en mujeres en el Registro Fabry en las siguientes edades:
%201.2023-10-09-14-24-34.svg)
La enfermedad de Fabry es progresiva: se debe controlar a las pacientes para detectar tempranamente las complicaciones potencialmente mortales.8-9
Se debe considerar la tre (terapia de reemplazo enzimático) ante la aparición de los primeros signos de enfermedad de fabry en mujeres
La TRE debe considerarse ante los primeros signos de complicaciones renales, cardíacas, del SNC (sistema nervioso central), dolor y alteraciones gastrointestinales.10-7
Pautas de consenso del panel internacional:10-7
Niños
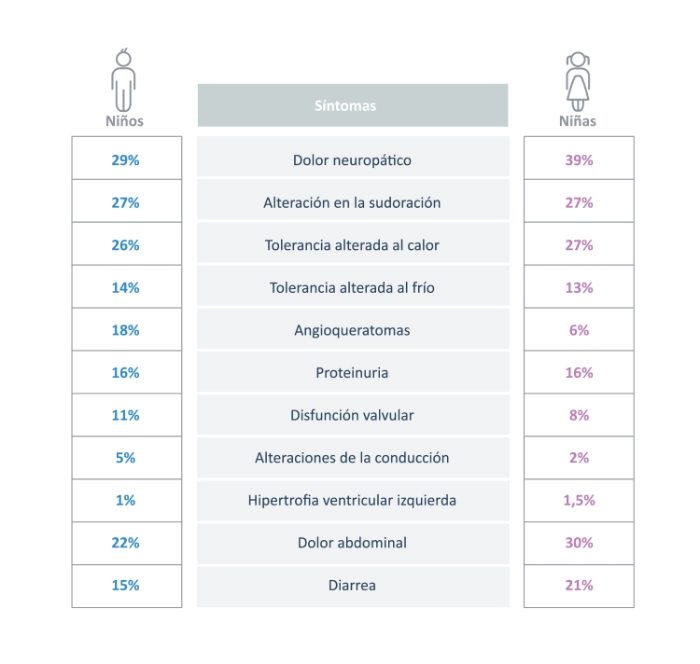
LOS NIÑOS CON ENFERMEDAD DE FABRY COMIENZAN A DESARROLLAR SÍNTOMAS A UNA EDAD TEMPRANA, QUE PROGRESAN A LO LARGO DEL TIEMPO11-12
Los niños con enfermedad de Fabry exhiben una amplia gama de síntomas:13
Las pautas recomiendan que los niños deben ser monitoreados de manera cercana14
Los niños pueden experimentar manifestaciones cardíacas y renales graves durante la infancia, y es importante asegurar la evaluación cercana de la progresión de la enfermedad para detectar los síntomas antes de que ocurra un daño irreversible11-13
La tre se debe considerar en los pacientes sintomáticos ≥ 8 años14
Signos y síntomas que justifican el tratamiento14
*En los estudios clínicos de Fabrazyme no se incluyeron pacientes menores a los 8 años de edad. No se ha evaluado la seguridad y eficacia de Fabrazyme en pacientes menores a los 8 años de edad.
La tre se debe considerar en los pacientes sintomáticos ≥ 8 años14
Signos y síntomas que justifican el tratamiento14
Hombres

La tre se debe considerar para los hombres al momento del diagnóstico6
Fabry clásico (sintomática o asintomática)6
-
La TRE debe considerarse y es apropiada
-
Las decisiones de tratamiento pueden estar influenciadas por la edad avanzada del paciente y la comorbilidad severa
Fabry no clásica o GLA VUS sin sentido*6
-
Se debe considerar la TRE y es apropiada si hay evidencia de laboratorio, histológica o de imágenes de lesión en el riñón, el corazón o el SNC, incluso en ausencia de los síntomas típicos de Fabry.
-
Las anormalidades deben ser atribuibles a la enfermedad de Fabry; esto puede requerir evaluación histológica o evidencia bioquímica de acumulación de GL-3
-
Se debe buscar el consejo de un experto en genética y manejo de la enfermedad de Fabry para la interpretación de la patogenicidad de cualquier VUS
El monitoreo regular es esencial para los hombres con la enfermedad de Fabry6
Se debe considerar la TRE y es apropiada si hay evidencia de laboratorio, histológica o de imágenes de lesión en el riñón, el corazón o el SNC, incluso en ausencia de los síntomas típicos de Fabry.
Las anormalidades deben ser atribuibles a la enfermedad de Fabry; esto puede requerir evaluación histológica o evidencia bioquímica de acumulación de GL-3
Se debe buscar el consejo de un experto en genética y manejo de la enfermedad de Fabry para la interpretación de la patogenicidad de cualquier VUS
*GLA VUS no clásico o sin sentido tiene la misma recomendación para hombres y mujeres.
- Krasnewich DM, Sidransky E. Chapter 208. In: Goldman-Cecil Medicine. 2016;1(25):1399-1403.
- Politei J, et al. Recomendaciones para el diagnóstico, tratamiento y seguimiento de la enfermedad de Fabry en Argentina. Revista Nefrología Argentina | ISSN 2591-278X Año 2018 | Edición Junio | Vol. 16 | Nro. 2
- Hopkin R, et al. Mol Genet Metab. 2016 Feb;117(2):104-13
- Politei J, et al. Pediatr Nephrol.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49.
- Barba-Romero MA, et al. Int J Clin Pract. 2011;65(8):903-910.
- Mehta A. QJM. 2002;95(10):647-653.
- Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112-128.
- Nagueh SF. Circulation. 2014;130(13):1081-1090.
- Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-1607.
- Sims K. et al. Stroke. 2009:40(3):788-794.
- The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed January 19, 2018.
- Wang RY et al. Genet Med 2007;9(1):34-45.
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-427.
Información relevante sobre cardiología
Miocardiopatías familiares
Las miocardiopatías familiares son enfermedades cardíacas hereditarias que afectan el miocardio, y por lo tanto tienen una base genética y requieren de un abordaje familiar1-5. Estás miocardiopatías se clasifican como hipertrófica (MCH)6-8, dilatada (MCD)9, restrictiva (MCR)10, no compactada (MCNC)11 y arritmogénica (MCA)12. A su vez, cada una de estas entidades engloban diversas enfermedades que tienen un alto nivel de complejidad, dado que son producidas por diferentes genes, tienen una gran heterogeneidad en la forma de expresión, sumado a una gran variabilidad en las manifestaciones clínicas y en el pronóstico. Es muy importante que los cardiólogos estudiemos estas familias mediante una profunda evaluación clínica, electrocardiográfica y la utilización de estudios de imágenes. Por otra parte, gracias a los grandes avances en genética cardiovascular, tenemos la posibilidad de sumar el enfoque molecular que nos brinda importante información para el diagnóstico, pronóstico y para llevar a cabo un cribado familiar más preciso. Actualmente sabemos que un mismo gen se puede expresar fenotípicamente como una MCH, MCD, MCR, MCNC y/o MCA1-12. En ocasiones también puede haber "fenotipos solapados, superpuestos o compartidos" y en un mismo paciente podemos encontrar por ejemplo criterios de MCH y MCNC asociados a una misma variante genética1-12. Las manifestaciones clínicas individuales y el pronóstico dependen de muchos factores además de la mutación causal primaria, y por esa razón cualquier variante genética tendrá presentaciones clínicas variables en diferentes individuos1-12. La información clínica sumada a los exámenes de laboratorio, estudios de imágenes y la identificación del defecto genético específico, nos ayudará a tener una mejor comprensión de los mecanismos involucrados en la expresión de la enfermedad y nos brinda más precisión para estratificar el riesgo de nuestros pacientes1-12. Algunos diagnósticos genéticos tienen implicaciones directas en pacientes que requieren terapias específicas que mejoran su pronóstico, como es el caso de la amiloidosis familiar13 y las enfermedades de Fabry14-15 o Pompe16-17. Las nuevas técnicas de secuenciación masiva en paralelo o ultrasecuenciación nos permiten identificar en un mismo paciente la presencia de más de una variante genética asociada a diferentes miocardiopatías u otras cardiopatías familiares18-19. Por lo tanto, la identificación de la etiología molecular específica de la enfermedad ayuda a orientar la utilización de las diferentes alternativas terapéuticas que disponemos actualmente, y es también la base para nuevas terapias que se encuentran actualmente en desarrollo13.
Utilidad práctica de la genética en las miocardiopatías familiares
En los últimos años hubo un progreso muy importante en la identificación de las causas genéticas de las miocardiopatías familiares1-17. También se ha avanzado de las técnicas de secuenciación tradicional Sanger a la secuenciación masiva en paralelo lo que ha permitido secuenciar todos los genes relacionados con una enfermedad en un sólo estudio, en un período corto de tiempo y con un costo menor18. Dado que este método brinda mucha información, es necesario contar con grupos multidisciplinarios especializados que dispongan de sistemas bioinformáticos adecuados y que la interpretación final de los hallazgos se lleve a cabo por médicos capacitados, preferentemente cardiólogos especializados en genética18. Dado la naturaleza heterogénea de las miocardiopatías familiares, el estudio genético es una herramienta muy útil para determinar cuál es la causa específica de la enfermedad e identificar enfermedades que en ocasiones se solapan clínicamente con miocardiopatías de otro origen (como ocurre por ejemplo en la enfermedad de Fabry)14-15. A pesar de que todas estas enfermedades son principalmente de origen hereditario, las pruebas diagnósticas actuales no permiten identificar las causas genéticas en un porcentaje variable de casos18-19. Hay que tener presente que un estudio genético negativo no descarta el diagnóstico ni la condición heredable de la enfermedad18 El estudio genético también es una herramienta muy útil para realizar un cribado genético familiar19Para ello, tenemos que elegir apropiadamente el primer paciente a estudiar genéticamente dentro de una familia "caso índice", prefiriéndose aquellos casos con cardiopatías más severas o con manifestaciones precoces de la enfermedad19. En el caso de que se logre identificar en el caso índice una o más variantes genéticas causantes de la enfermedad, luego de un asesoramiento genético especializado se puede hacer un estudio en cascada en los familiares de primer grado para determinar si son portadores de la o las variantes genéticas19. La información que se obtenga en el resto de la familia ayudará a pesquisar quienes están predispuestos a desarrollar la enfermedad y determinar quienes no son portadores de la alteración genética y por lo tanto no necesitarán un seguimiento rutinario18-19. Por último, en algunas familias la genética nos puede brindar información pronóstica adicional. Sin embargo, no debemos olvidar que el estudio genético es un método complementario más, y que nunca debemos decidir una conducta terapéutica basándonos solamente en este aspecto, sin previamente realizar una adecuada evaluación y estratificación de riesgo clínica18-19.
Información relevante sobre neurología
Accidente cerebrovascular en jóvenes
Se considera accidente cerebrovascular-ACV- (o stroke) en pacientes jóvenes cuando se presenta en menores de 45 años. Se ha reportado una incidencia de 10 en 100,000 habitantes y una mortalidad del 2.5%. Si bien ante un ACV, hay una mejor supervivencia en pacientes jóvenes que en aquéllos de edad avanzada; sin embargo, la mayoría de jóvenes sobrevivientes tienen secuelas emocionales, sociales y/o físicas que deterioran su calidad de vida.
Los factores de riesgo más importantes asociados a la presencia de ACV en jóvenes son el tabaquismo, la dislipidemia, cardiopatía, diabetes mellitus, hipertensión arterial y antecedentes familiares de ACV en jóvenes en la misma familia.
Existe un creciente reporte de casos asociados al consumo de drogas; se describe que el uso de anfetaminas, cocaína y otras drogas excitatorias causan vasoespasmo y posible oclusión de la arteria cerebral media; sin embargo, el uso de cocaína está relacionado tanto a eventos isquémicos como hemorrágicos. El embolismo cardiaco (cardiopatía reumática, la persistencia del foramen oval y el prolapso mitral) es la causa más frecuente de ACV en pacientes menores de 40 años, mientras que la aterosclerosis es más frecuente entre los 41 y 50 años. Estos resultados en relación a la incidencia pueden variar en algunas series dependiendo el país que la reporta.
Los trastornos de la coagulación son otras causas frecuentes en la población joven respecto a los adultos y ancianos. Las más frecuentes a estudiar son: la eficiencia de la proteína C, deficiencia de la proteína S, resistencia a la proteína C, deficiencia de la antitrombina III, deficiencia del plasminógeno, síndrome antifosfolípidos, paraproteinemias y relacionadas al uso de anticonceptivos orales.
Un grupo de enfermedades genéticas ha sido incluido como causas a investigar en pacientes jóvenes con ACV, siendo las más comunes: la arteriopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía (CADASIL), la endoteliopatía hereditaria asociada a retinopatía, nefropatía e ictus (HERNS), enfermedad de Fabry y la encefalopatía mitocondrial con acidosis láctica e ictus (MELAS).20-21
Dolor neuropático en pacientes jóvenes
El dolor neuropático es el dolor proveniente de una lesión o enfermedad que afecta al sistema somatosensitivo central o periférico. Suele ser el marcador en muchos casos de una neuropatía de fibras finas (o de pequeño calibre). Dala la complejidad de sus síntomas y de su fisiopatología, incluso con la evaluación apropiada, entre un 25% y un 40% de las neuropatías de fibras finas permanecerán sin causa definida.
Las principales manifestaciones de las neuropatías de fibras finas son la sensación quemante en los pies (a veces concomitantemente en las manos) y la alteración de la sensibilidad térmica y dolorosa. La sensibilidad epicrítica y proprioceptiva es normal, como también los reflejos osteomusculares, ya que esas manifestaciones se asocian a las neuropatías de fibras gruesas.
Las manifestaciones sensitivas “negativas” son la hipoalgesia y la hipoestesia y las “positivas” son parestesia, disestesia, hiperpatía, hiperalgesia y alodinia, además de las sensaciones de prurito y hormigueo. La fisiopatología del dolor neuropático es compleja y envuelve los siguientes mecanismos: Sensibilización de nociceptores; activación espontánea de las fibras aferentes y de los nociceptores; regulación ascendente disfuncional de los canales de sodio (Nav 1.3, Nav 1.7, Nav 1.8, Nav 1.9), y descargas ectópicas del ganglio de la raíz dorsal entre otros.
Las causas más comunes que derivan en una neuropatía de fibras finas son: la diabetes, la insuficiencia renal, algunos fármacos, la deficiencia de vitaminas, las infecciones por VIH y hepatitis C, enfermedades autoinmunes, intoxicaciones por metales pesados, canalopatías genéticas, síndromes paraneoplásicos y enfermedades por depósito lisosomal (ej: enfermedad de Fabry).
El diagnóstico se basa en el examen físico, estudios serológicos y de laboratorio específicos, test neurofisiológicos (test de cuantificación sensitiva) y en algunos casos la biopsia de piel con cuantificación de terminales libres.
Actualmente la realización de un familiograma o árbol genealógico se considera relevante en los pacientes que presentan dolor neuropático en crisis (o paroxístico). La presencia de este tipo de dolor quemante en crisis en los cuatro miembros nos acerca a la posibilidad de una canalopatía primaria (mutaciones en genes que codifican proteínas asociadas a los canales de sodio) o secundaria (depósito lisosomal de un sustrato que resulta en daño ganglionar y de fibras finas como en la enfermedad de Fabry).
El tratamiento deberá ajustarse a la causa determinada. Al mismo tiempo existen una gran variedad de fármacos que por medio del bloqueo de distintos receptores (canales de sodio, de calcio, inhibidores de la recaptación de neurotransmisores, etc) pueden resultar en un alivio de los síntomas dolorosos.22-24
Información relevante sobre dermatología
Genodermatosis. la piel como clave para el diagnóstico de enfermedades genéticas
Las genodermatosis son un grupo heterogéneo de enfermedades de la piel y los anexos cutáneos (pelo y uñas) que se caracterizan por tener un origen genético. Las manifestaciones cutáneas pueden estar presentes desde el nacimiento o manifestarse más tarde, durante el desarrollo. Las mismas pueden ser especificas o inespecíficas, pero en muchas oportunidades son la puerta de entrada al diagnóstico de síndromes con manifestaciones extra cutáneas ya que la piel es un órgano sumamente accesible para la realización de una biopsia o el reconocimiento de patrones clínicos que pueden orientar a un diagnóstico.
A los fines prácticos se pueden dividir a las genodermatosis según diversos criterios.
Criterio clínico:
Síndromes neuro-cutáneos: Son aquellos en los que predominan los síntomas dermatológicos y neurológicos. Ej: Neurofibromatosis, esclerosis tuberosa e incontinencia pigmenti, etc.
Genodermatosis con potencial maligno: Son aquellos en los que se asocian manifestaciones cutáneas y tendencia a desarrollar tumores (cutáneos o extra-cutáneos) Ej: Xeroderma pigmentoso, Sme de Brooke Spiegler, Sme de Cowden, Sme de Gorlin, disqueratosis congenita, etc.
Síndromes autoinflamatorios monogénicos: Son enfermedades genéticas que afectan a los genes que regulan la inflamación. Sme TRAPS, Sme CINCA/NOMID/, Sme CANDLE, Sme DIRA/DITRA, Sme SAVI.
Criterio Clínico - Dermatológico:
Divide a las dermatosis según su fenotipo cutáneo, independientemente de si tienen o no compromiso extra cutáneo o cuál sea el gen responsable.
Ictiosis: Ictiosis ligada al X, ARCI, KID, Gaucher, etc.
Queratodermia palmoplantar (QD): QD de Unna-Thost, enfermedad de Maleda, QD de Vörner, sme de Papillon-Lefevre.
Epidermólisis ampollar (EA): EA simple, EA distrófica, EA de la unión etc.
Displasias ectodérmicas (DE): DE anhidriotica, DE hidrotica, Sme de Basan etc.
Genodermatosis con fotosensibilidad: Porfirias, Xeroderma pigmentoso, Sme de Cockayne, etc.
Angioqueratoma corporal difuso: Enfermedad de Fabry, Fucosidosos, Beta Manosidosis, alfa-galactosidosis, galactosialidosis.
Mancha mongólica aberrante y progresiva: Mucopolisacaridosis I, II, VI, Mucolipidosis, etc.
Acrodermatitis enteropatica-simil: Acidemia metilmalonica, acidemia propionica, enfermedad de la orina en jarabe de arce, fenilcetonuria, deficiencia de Ornitil transcarbamilasa y citrulinemia.
Criterio Genético - fisiopatogénico
RASOPATIAS: aquellas enfermedades que presentan mutaciones en las vías RAS/MAP. NF1, Sme de Noonan, Sme de Legius, Sme de Costello, CFC, Sme de LEOPARD.
Síndromes PROS (Síndromes por sobrecremiento asociados a PIK3CA): CLOVES, hemihiperplasia con lipomatosis múltiple, hiperplasia fibroadiposa.
Como en todas las enfermedades genéticas, el interrogatorio exhaustivo prestando especial atención a los antecedentes familiares, la presencia de síntomas similares en los padres, el antecedente de abortos o de múltiples signos y síntomas que no parecieran estar relacionados en distintos miembros de la familia y un examen físico completo, nos puede orientar a la presencia de una genodermatosis y orientaros a la solicitud de estudios complementarios acordes (biopsia de piel, estudios por imágenes, laboratorios de rutina o específicos, como dosajes hormonales u enzimáticos e incluso estudios genéticos).
En la actualidad el diagnóstico temprano de muchas de estas enfermedades implica la posibilidad de instaurar tratamientos específicos que pueden modificar la historia natural o al menos realizar intervenciones preventivas o controles específicos a fines de prevenir complicaciones a largo plazo, por eso la importancia de estar alertas 25-25.
Información relevante sobre oftalmología
La oftalmología como herramienta para detectar enfermedades genéticas
La oftalmología juega un rol preponderante en la genómica humana28, lo que incluye la terapia génica. Es el cuarto órgano más comúnmente afectado luego de tegumento (piel, cabello y uñas), sistema nervioso y sistema musculoesquelético en las enfermedades genéticas29.
El ojo está involucrado en trastornos monogénicos y en aquellos causados por etiología multifactorial. Como ejemplos se encuentran: la neuropatía óptica hereditaria de Leber, que fue la primera descripta en la literatura como trastorno mitocondrial y la ceguera para el color rojo/verde fue la primera enfermedad recesiva ligada al X descripta. A diferencia de cualquier otro órgano tiene la enorme ventaja de permitirnos la visualización directa de fenómenos genéticos tales como la inactivación sesgada del cromosoma X en el fondo de ojo de una mujer portadora de albinismo ocular30.
El campo de la genética está evolucionando a un ritmo acelerado31.
Casi el 50% de la ceguera pediátrica se debe a una etiología genética. La afectación ocular es superada en frecuencia solo por el cerebro como órgano afectado de manera individual.
Los conceptos de genética y su implicancia en la clínica oftalmológica son importantes, ya que conociéndolos se puede lograr un manejo adecuado y un asesoramiento genético oportuno a pacientes y sus familiares.
Una parte crítica de una evaluación genética es el árbol genealógico (pedigrí).
Típicamente, se construyen al menos tres generaciones. Su importancia radica en proveer información sobre el patrón de la herencia, etnia, los antecedentes familiares; nos ayuda también a identificar familiares en potencial riesgo de padecer enfermedad hereditaria, logra que establezcamos una buena relación médico/paciente (clave en toda enfermedad y más aún en trastornos crónicos), nos permite brindar consejería respecto a planificación familiar, y en fin es muy importante tenerlo ya que una vez constituido forma parte de la historia clínica del paciente y es de buena práctica médica.
Para concluir el primer paso siempre será realizar un examen oftalmológico básico en todos los pacientes, y en algunos requerirán también estudios tales como: la lámpara de hendidura que tiene como función el análisis de la porción anterior del ojo, el fondo de ojo que nos sirve para observar la retina, sus vasos sanguíneos, la papila óptica y la macula, entre otros estudios complementarios oftalmológicos.
Información relevante sobre pediatría32-36
Los desórdenes gastrointestinales funcionales (DGF) abarcan un amplio rango de trastornos atribuibles al tracto gastrointestinal que no pueden ser explicados por anormalidades estructurales o bioquímicas.
Síndrome de rumiación
En el síndrome de rumiación infantil, los bebés regurgitan habitualmente y voluntariamente el contenido del estómago en la cavidad oral como una salida de conducta autoestimuladora, asociado a una privación social de larga data. Puede causar desnutrición.
Síndrome de vómito cíclico (CVS)
Episodios recurrentes de vómitos que duran de horas a días con períodos de intervalo sin síntomas que duran semanas o meses. El tratamiento tiende a prevenir episodios frecuentes, severos y prolongados.
Colicos infantiles
Se caracterizan como períodos recurrentes y prolongados de llanto, alboroto o irritabilidad, sin causa obvia y no resuelto o prevenibles por los cuidadores, sin evidencia de falta de desarrollo, fiebre o enfermedad en un bebé menor de 5 meses al inicio y resolución de los síntomas. El microbioma sería importante en la fisiopatogenia.
Diarrea funcional
A pesar de tener frecuentes deposiciones desligadas, no presentan trastornos en el crecimiento, siempre que la ingesta calórica sea adecuada. Los síntomas resuelven en la edad escolar. Existen factores dietarios involucrados en la fisiopatología de esta entidad. Se recomienda corregir la dieta.
Disquecia del lactante
Se caracteriza por esfuerzo, gritos, llanto y coloración purpura de la cara, al hacer esfuerzo defecatorio, en un lactante con deposiciones blandas. El mecanismo sería una falta de coordinación entre el aumento de la presión abdominal y los músculos del piso pélvico al defecar .Diferenciarla de constipación. Resolución espontanea
Constipación funcional
Los síntomas incluyen heces duras, movimientos intestinales dolorosos y en niños pequeños incontinencia fecal. Experiencias defecatorias poco placenteras (defecación dolorosa), pueden causar retención voluntaria para evitar una situación displacentera . La retención origina una reabsorción aumentada de agua de las heces, que se endurecen, generando dolor y dificultad evacuatoria. Ante un bolo fecal, se debe desimpactar, seguido de un tratamiento de mantenimiento con laxantes. En ambas instancias, se recomienda el polietilenglicol.
Dolor
Es un componente importante de muchas DGF en neonatos y niños pequeños. El modelo tradicional de dolor agudo no es adecuado para el manejo del dolor funcional. El dolor funcional tiene en cuenta la influencia de otros elementos en la interpretación y respuesta a la información nociceptiva, como factores psicosociales, factores ambientales, predisposición genética y sistemas reguladores del dolor deteriorados. En la vida temprana, el umbral de dolor es bajo y aumenta con la edad. Se combina con una disminución del control inhibitorio para la modulación de experiencias de dolor. En el largo plazo, las experiencias dolorosas en recién nacidos pueden resultar en alteraciones prolongadas en los mecanismos de procesamiento del dolor, que pueden resultar en hiperalgesia visceral a una edad más temprana. Esto se considera un mecanismo fisiopatológico importante subyacente dolor abdominal funcional.
Evaluación del dolor
Los niños más pequeños, utilizan escalas de clasificación modificadas con caras doloridas. Los bebés y la mayoría de los niños pequeños no son capaces de escalar su dolor. La evaluación de factores fisiológicos (frecuencia cardíaca, presión arterial, saturación de oxígeno) y comportamientos asociados con el dolor (expresiones faciales, comportamiento motor, llanto), se han desarrollado para apoyar al clínico.
Información relevante sobre nefrología
Calculadora de filtrado Glomerular
Nefrogenética: aplicación de conceptos básicos de genética a las enfermedades renales para el Nefrólogo Clínico37-41
Los avances en genética y biología molecular impulsaron la investigación de las enfermedades renales hereditarias y el trabajo conjunto de Nefrólogos y Genetistas para la asistencia clínica y el asesoramiento genético adecuado de los pacientes que las padecen. Así fue como los avances en genética molecular se comenzaron a aplicar a las enfermedades renales y surgió el concepto de Nefrogenética en la década de 1980.
Los avances en el campo de la genética molecular han incluso, cambiado considerablemente la clasificación de las enfermedades renales glomerulares o quísticas hereditarias.
El creciente progreso de la Biología molecular ha significado una repercusión importante en diversos aspectos de la nefrología clínica:
- El descubrimiento de nuevas mutaciones genéticas causantes de enfermedades renales ha permitido a los nefrólogos conocer el pronóstico, tanto renal como extra-renal de ciertas nefropatías.
- Se han logrado modelos de enfermedades renales en animales, tanto espontáneas como inducidas genéticamente, de modo que se asemejen a las nefropatías en humanos, para su mejor conocimiento.
- El descubrimiento de dianas terapéuticas de ciertas nefropatías ha significado importantes avances en los tratamientos específicos de algunas nefropatías de causa genética. Algunos ejemplos de ello son la poliquistosis renal de origen autosómico, la deficiencia en la actividad de la adenina fosforribosiltransferasa, la cistinosis y la enfermedad de Fabry, entre otras.
Las enfermedades genéticas se consideran, en su gran mayoría, enfermedades raras, por su baja prevalencia individual. Se estima que el 10-20% de los adultos con enfermedad renal crónica tienen una enfermedad genética hereditaria de base, aunque esta cifra debe ser considerada inferior a la real ya que las enfermedades genéticas subyacentes están infra-diagnosticadas. Las ER suelen ser difíciles de diagnosticar y es por eso que el concepto de trabajo conjunto entre Genetistas y Nefrólogos debe ser reforzado.
Enfoque práctico de la proteinuria en la enfermedad renal42-46
La proteinuria es un bio-marcador conocido de daño renal, un importante factor de progresión en la enfermedad renal crónica (ERC) y un potente predictor de eventos cardiovasculares y mortalidad en pacientes con nefropatía crónica de diversas causas. Aunque no es específica de enfermedad glomerular, una proteinuria anormalmente elevada es una manifestación principal en la mayoría de los pacientes con glomerulopatías.
Para la cuantificación adecuada de la excreción de proteínas urinarias existen, en la actualidad, protocolos validados tanto para la recogida de la muestra, como para las técnicas de laboratorios utilizadas (etapa preanalítica y analítica), así como valores de referencia y además algunas controversias respecto a la utilidad de la determinación de proteinuria o albuminuria.
La primera etapa y la más importante en la evaluación de un paciente con proteinuria es el desarrollo de un diagnóstico diferencial adecuado, debido a que una excreción urinaria aumentada de proteínas urinarias se produce por procesos tanto fisiológicos como patológicas y, entre estos últimos, las causas pueden ser diversas. La anamnesis sobre antecedentes tanto familiares como personales, hábitos, transfusiones, uso exposición a fármacos, ciertos signos o síntomas claves del interrogatorio y del examen físico deben ser priorizados al momento de la evaluación del paciente con proteinuria que suele presentarse a la consulta con síndrome nefrótico en las proteinurias intensas, como principal manifestación.
Una adecuada evaluación inicial del paciente con proteinuria permitirá al Nefrólogo Clínico la solicitud racional de exámenes complementarios y un enfoque adecuado de tratamiento.
La proteinuria puede obedecer a enfermedades frecuentes como la Diabetes Mellitus o Hipertensión Arterial, así como a enfermedades glomerulares de origen autoinmune o a causas menos frecuentes hereditarias, como la enfermedad de Fabry.
- Barriales-Villa, Gimeno-Blanes, Zorio-Grima, Ripoll-Vera, Evangelista-Masip, Moya-Mitjans, Serratosa-Fernández, Albert-Brotons, García-Pinilla, García-Pavía. Plan of Action for Inherited Cardiovascular Diseases: Synthesis of Recommendations and Action Algorithms. Rev Esp Cardiol 2016;69(3):300-9. doi: 10.1016/j.rec.2015.11.029.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006 Apr 11;113(14):1807-16. DOI: 10.1161/CIRCULATIONAHA.106.174287.
- Perry Elliott, Bert Andersson, Eloisa Arbustini, Zofia Bilinska, Franco Cecchi, Philippe Charron, Olivier Dubourg, Uwe Kühl, Bernhard Maisch, William J. McKenna, Lorenzo Monserrat, Sabine Pankuweit, Claudio Rapezzi, Petar Seferovic, Luigi Tavazzi, Andre Keren. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008;29:270-6. doi.org/10.1093/eurheartj/ehm342 .
- Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 2013 Dec 3;62(22):2046-72. doi: 10.1016/j.jacc.2013.08.1644. Epub 2013 Nov 18. No abstract available. Erratum in: J Am Coll Cardiol 2014 Jan 21;63(2):191-4.
- Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L, Tendera M, Maupain C, et al. EORP Cardiomyopathy Registry Investigators. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J 2018;39(20):1784-1793. doi: 10.1093/eurheartj/ehx819.
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2011 Dec 13;58(25):2703-38. doi: 10.1016/j.jacc.2011.10.825..
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Authors/Task Force members, Eur Heart J 2014;35(39):2733-79. doi: 10.1093/eurheartj/ehu284.
- Fernández A, Acunzo RS, Avegliano G, Casabé JH, Dumont CA, Hita A, Ortiz M, Pérez de Arenaza D, y col. Consenso Argentino de diagnóstico y tratamiento de la Miocardiopatía Hipertrófica 2016. Rev Argent Cardiol 2017;85 (Suplemento 2):1-78.
- Cuenca Cuenca S, Ruiz-Cano MJ, Gimeno-Blanes JR, Jurado A, Salas C, Gomez-Diaz I, Padron-Barthe L, Javier Grillo J, et al. Inherited Cardiac Diseases Program of the Spanish Cardiovascular Research Network (Red Investigación Cardiovascular). Genetic Basis of Familial Dilated Cardiomyopathy Patients Undergoing Heart Transplantation J Heart Lung Transplant 2016;35(5):625-35. doi: 10.1016/j.healun.2015.12.014.
- Gallego-Delgado M, Delgado JF, Brossa-Loidi V, Palomo J, Marzoa-Rivas R, Perez-Villa F, Salazar-Mendiguchía J, Ruiz-Cano MJ, et al. Idiopathic Restrictive Cardiomyopathy Is Primarily a Genetic Disease Heart Rhythm 2019;16(11):e373-e407. doi: 10.1016/j.hrthm.2019.09.019J Am Coll Cardiol.
- Yvonne M Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, ten Cate FJ, et al. The Importance of Genetic Counseling, DNA Diagnostics, and Cardiologic Family Screening in Left Ventricular Noncompaction Cardiomyopathy Circ Cardiovasc Genet 2010;3(3):232-9. doi: 10.1161/CIRCGENETICS.109.903898. Epub 2010 Jun 8.
- Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, et al. HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 2019;16(11):e373-e407. doi: 10.1016/j.hrthm.2019.09.019.
- Giuliana G. Repetti, Christopher N. Toepfer, Jonathan G. Seidman, Christine E. Seidman. Novel Therapies for Prevention and Early Treatment of Cardiomyopathies: Now and in the Future. Circ Res 2019; 124(11): 1536–1550. doi: 10.1161/CIRCRESAHA.119.313569.
- Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, Eng C, Hopkin RJ, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 2018 Apr;123(4):416-427. doi: 10.1016/j.ymgme.2018.02.014.
- Politei J, Aiziczon D, Aguilar M, Alberton V, Alonso S, Amoreo O, Andrade L, Antongiovanni N, et al. Recomendaciones para el diagnóstico, tratamiento y seguimiento de la enfermedad de Fabry en Argentina. Revista Nefrología Argentina 2018;16(2):1-29.
- Dubrovsky A, Fulgenzi E, Amartino H, Carlésd D, Corderi J, de Vito F, Fainboimg A, Ferradásh N, et al. Consenso Argentino para el diagnóstico, seguimiento y tratamiento de la Enfermedad de Pompe. Neurolarg 2014; 6: 96-113
- Dubrovsky A, Fulgenzi E, De Vito EL, Barroso F, Berardo A, Bettini M, Binaghi D, Calabrese E, et al.Consenso Argentino sobre enfermedad de Pompe de inicio tardío. Medicina (B Aires) 2018;78 Suppl 1:1-23.
- Monserrat L, Ortiz-Genga M, Lesende I, Garcia-Giustiniani D, Barriales-Villa R, de Una-Iglesias D, Syrris P, Castro-Beiras A. Genetics of cardiomyopathies: novel perspectives with next generation sequencing. Curr Pharm Des 2015;21(4):418-30. doi: 10.2174/138161282104141204123748.
- Charron, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, et al. European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31(22):2715-26. doi: 10.1093/eurheartj/ehq271.
- Gupta S, et al. Medicine (Baltimore). 2005;84(5):261-268.
- Shelley ED, et al. Pediatr Dermatol 1995;12(3):215-219. 19. Fabry Registry Annual Report 2010. Available at: www.fabry.org/fsig.nsf/PDFs/PDFsR/$File/2010_Annual_Report.pdf. Accessed January 22. 2018.
- Ramaswami U, et al. Clin J Am Soc Nephrol. 2010;5(2):365-370.
- Ichinose M, et al. Clin Exp Nephrol. 2005;9(3):228-232 .
- Bekri S, et al. Nephrol Clin Pract 2005:101(1):c33-38.
- Coresh J, et al. JAMA. 2007;298(3):2038-2047.
- Waldek S, et al. BMC Nephrol. 2014:15(72):1-15.
- Najafian B, et al. Kidney Int. 2011;79(6):663.670.
- Torra R. Kidney Int. Suppl. 2008;(Suppl. 111):S29-S32.
- Schiffmann R, et al. Nephrol Dial Transplant. 2009.24(7) 2102-2111.
- Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505.517.
- Linthorst GE, et al. J Med Genet. 2010;47(4):217-222.
- Monserrat L, et al. Coll Cardiol. 2007;50(25):2399-2403.
- van der Tol L, et al. J Med Genet. 2014;51(1):1-9.
- Yousef Z, et al. Eur Heart J. 2013;34(11):802-808.
- Eng CM, et al. Genet Med. 2006;8(9):539-548.
- Linhart A, et al. Eur Heart J. 2007;28(10):1228-1235.
- Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-1099.
- Kampmann C, et al. Int J Cardiol. 2008:130(3):367-373.
- Weidemann F, et al. Orphanet J Rare Dis. 2013:8(116).
- Elliott PM, et al. Eur Heart J. 2014;35(39):2733-2779.
- Fellgiebel A, et al. Lancet Neurol. 2006;5(9):791-795.
- Hilz MJ. Clin Ther. 2010;32 (suppl C):S93.
- Bottcher T. et al. Plos ONE. 2013:8(8):e71894.
- Cable WJL, et al. Neurology. 1982;32(5):498-502.
- Uceyler N, et al. Clin J Pain. 2014;30(10):915-920.
- Fellgiebel A, et al. Neurology. 2009;72(1):63-68.
- Moore DF, et al. Brain Res Bull. 2003;62(3):231-240.
- Cole AL, et al. J Inherit Metab Dis. 2007:30(6):943-951.
- Samiy N, et al. Surv Ophthalmol. 2008;53(4):416-423.
- Hopkin RJ, et al. Pediatr Res. 2008;64(5):550-555.
- MacDermot KD, et al. J Med Genet. 2001a;38(11):750-760.
- MacDermot KD, et al. J Med Genet. 2001b;38(11):769-775.
- Manger B, et al. Clin Rheumatol. 2007;26(3):335-341.
- Ekker MS, et al. Epidemiology, aetiology, and management of ischaemic stroke in young adults. Lancet Neurol. 2018 Sep;17(9):790-801.
- González-Gómez FJ, et al. Stroke in young adults: Incidence rate, risk factors, treatment and prognosis. Rev Clin Esp. 2016 Oct;216(7):345-351.
- Walco GA, et al. Neuropathic pain in children: Special considerations. Mayo Clin Proc. 2010 Mar;85(3 Suppl):S33-41 .
- Durval Campos Kraychete, et al. Neuropatías Periféricas Dolorosas. Rev Bras Anestesiol 2011; 61: 5: 351-360 .
- Görlach J, et al. Diagnostic utility of small fiber analysis in skin biopsies from children with chronic pain. Muscle Nerve. 2020 Feb;61(2):173-181.
- Genetic skin disorders. Virginia Sybert. Oxford University Press, ED. 2010. 26 Luna PC, Abad ME, Boggio P, et al. Enfermedades por depósito lisosomal. Diagnóstico a partir de lesiones cutáneas. Dermatología Argentina, Vol 17, No 3 (2011).
- Hernández-Martín A, Torrelo A. Rasopatías: trastornos del desarrollo con predisposición al cáncer y manifestaciones cutáneas. Actas Dermatosifil 2011;102:402-416.
- Wygnanski-Jaffe T, Levin AV. Introductory genetics for the ophthalmologist. American Academy of Ophthalmology, Focal Points. Clin Module Ophthalmol 2005;23:1-11.
- Costa T, Scriver CR, Childs B. The effect of Mendelian disease on human health: A measurement. Am J Med Genet 1985;21:231-42.
- Sadagopan KA, Capasso J, Levin AV. Genetics for the ophthalmologist. Oman J Ophthalmol. 2012;5(3):144‐149. doi:10.4103/0974-620X.106092.
- Committee on Bioethics. Ethical issues with genetic testing in pediatrics. Pediatrics 2001;107:1451.
- Benninga MA ,et al.Childhood Functional Gastrointestinal Disorders: Neonate/Toddler. Gastroenterology. May 2016 Volume 150, Issue 6, Pages 1443–1455.e2.
- Baaleman DF, et al. The Effects of the Rome IV Criteria on Pediatric Gastrointestinal Practice. Curr Gastroenterol Rep. 2020 Mar 19;22(5):21.
- Edwards T, et al. Classification of pediatric functional gastrointestinal disorders related to abdominal pain using Rome III vs. Rome IV criterions. BMC Gastroenterol. 2018 Mar 17;18(1):4.
- Rajindrajith S, et al. Functional abdominal pain disorders in children Expert Rev Gastroenterol Hepatol. 2018 Apr;12(4):369-390.
- Zeevenhooven J, et al. Definitions of Pediatric Functional Abdominal Pain Disorders and Outcome Measures: A Systematic Review. J Pediatr. 2019 Sep;212:52-59.e16.
- Grünfeld, J. P., & Grünfeld, J. P. (2011). Nefrogenética: los genes, las enfermedades y los pacientes y sus familiares. Nefrología (English Edition), 2(1), 1-2.
- Guillen-Navarro, E., Ballesta-Martínez, M. J., & López-González, V. (2011). Genética y enfermedad. Concepto de genética médica. Nefrología, 2(1), 3-10.
- Hildebrandt, F. (2010). Genetic kidney diseases. The Lancet, 375(9722), 1287-1295.
- Marin, E. P., Cohen, E., & Dahl, N. (2020). Clinical Applications of Genetic Discoveries in Kidney Transplantation: A Review. Kidney360, 1(4), 300-305.
- Groopman, E. E., Rasouly, H. M., & Gharavi, A. G. (2018). Genomic medicine for kidney disease. Nature Reviews Nephrology, 14(2), 83.
- Avendaño, L. H. (2009). Nefrología clínica. Ed. Médica Panamericana. Schrier, R. W. (2018). Manual de nefrología. Thieme Revinter Publicações LTDA.
- Palmer, S. C., Ruospo, M., Teixeira-Pinto, A., Craig, J. C., Macaskill, P., & Strippoli, G. F. (2018). The validity of drug effects on proteinuria, albuminuria, serum creatinine, and estimated GFR as surrogate end points for ESKD: a systematic review. American Journal of Kidney Diseases, 72(6), 779-789.
- Currie, G., Taylor, A. H., Fujita, T., Ohtsu, H., Lindhardt, M., Rossing, P., ... & van den Meiracker, A. H. (2016). Effect of mineralocorticoid receptor antagonists on proteinuria and progression of chronic kidney disease: a systematic review and meta-analysis. BMC nephrology, 17(1), 127.
- Floege, J., Barbour, S. J., Cattran, D. C., Hogan, J. J., Nachman, P. H., Tang, S. C., ... & Rovin, B. H. (2019). Management and treatment of glomerular diseases (part 1): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney international, 95(2), 268-280.
- Rovin, B. H., Caster, D. J., Cattran, D. C., Gibson, K. L., Hogan, J. J., Moeller, M. J., ... & Floege, J. (2019). Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney international, 95(2), 281-295.
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.