
Descubriendo la enfermedad de Gaucher
La EG es una de las enfermedades lisosomales más frecuentes.
Se hereda de forma autosómica recesiva. Mutaciones en el gen de la beta-glucosiodasa (GBA1)
Prevalencia:
- 1 en 40.000-100.000 en la población general
- 1 en 800 en la población judío Ashkenazi
Temas cubiertos en esta sección:
Fisiopatología:
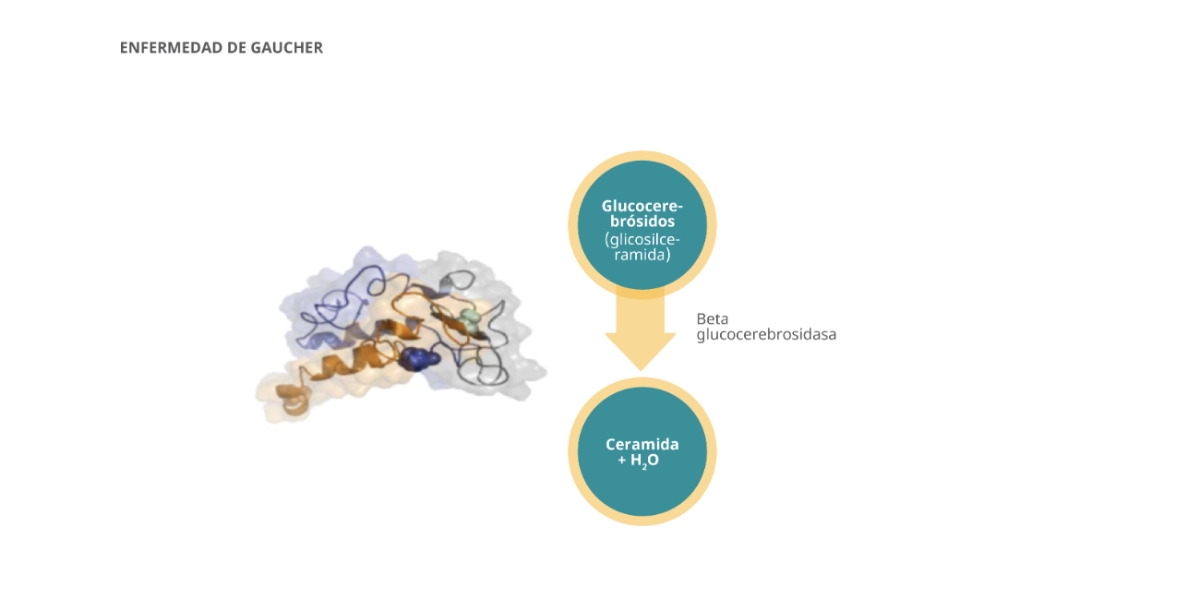
La enfermedad de Gaucher es una enfermedad metabólica hereditaria debida a un déficit de la enzima lisosomal β-glucosidasa ácida1.
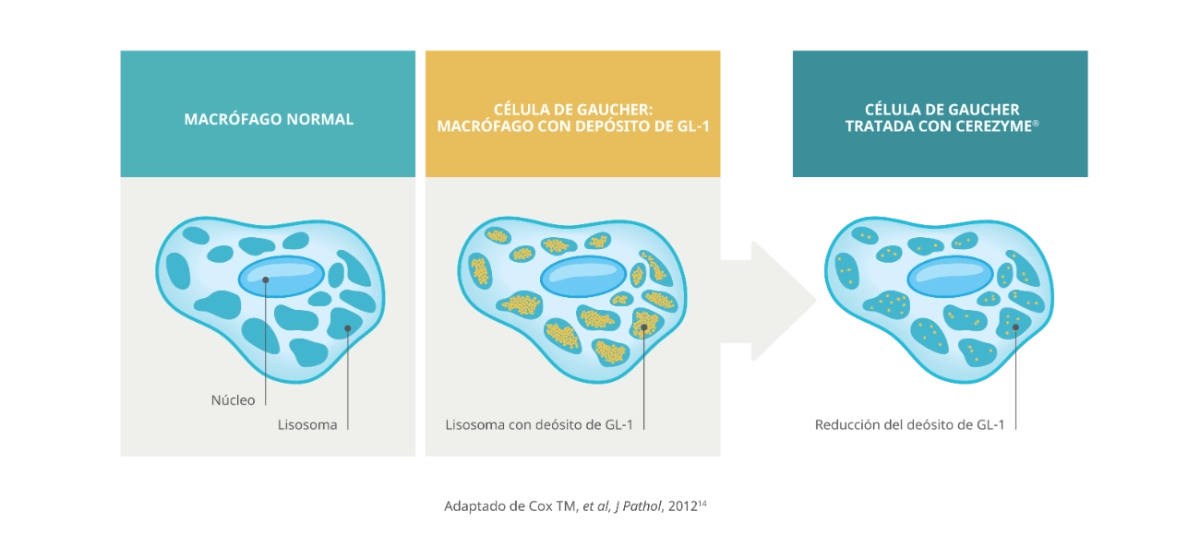
- En circunstancias normales, la β-glucosidasa ácida descompone la glucosilceramida (GL-1), un componente fundamental de la bicapa lipídica de las membranas celulares, en glucosa y ceramida.1
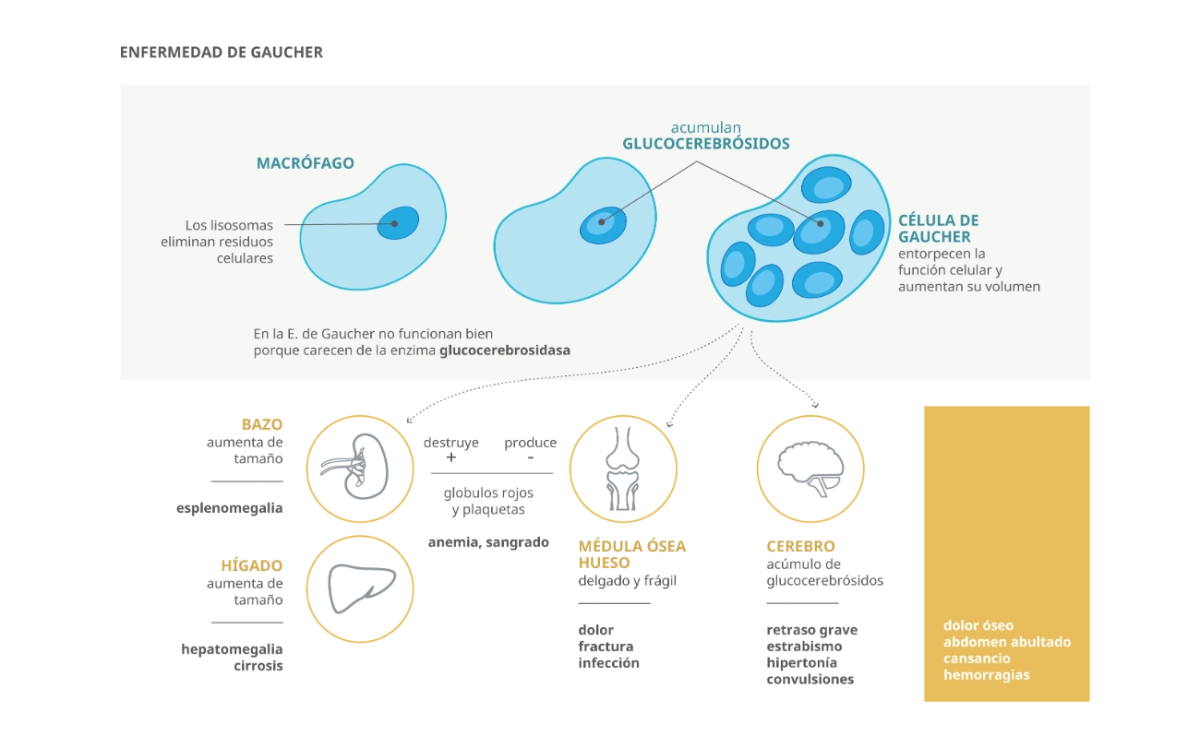
- En las personas con enfermedad de Gaucher, el déficit de actividad de la β-glucosidasa ácida provoca la acumulación de GL-1 en el interior de los lososomas de los macrófagos (células de Gaucher), especialmente en el bazo, el hígado y la médula ósea.1
Fisiología
(Gcase y glucocerebrósidos)
Cuadro clínico
La enfermedad de Gaucher es una patología crónica, progresiva y multisistémica.
La acumulación de macrófagos alterados en los distintos tejidos, puede provocar:
- Hepatomegalia
- Esplenomegalia
- Plaquetopenia, anemia
- Dolor óseo
- Neuropatía
- Afección pulmonar
Enfermedad de Gaucher
Enfermedad de Gaucher: formas clínicas
Otros alteraciones clínicas menos frecuentes:
- Infartos esplénicos y hepáticos: Crisis de dolor abdominal
- Gaucheroma hasta en 40% de los pacientes
- Lesiones focales en hígado y bazo
- Litiasis biliar: 5 veces más frecuente que población gral.
- Hipergammaglobulinemia policlonal o monoclonal
- Disminución de protrombina y TTPA
Formas clínicas
El cuadro clínico puede ser heterogéneo.
Existen 3 tipos distintos de Enfermedad de Gaucher:
Tipo 1: Forma No Neuropática. 95% de los casos
- Hepatoesplenomegalia leve a severa
- Citopenias: Anemia, trombocitopenia
- Compromiso óseo
- Compromiso pulmonar
- Mutación N370S es la más comúnmente asociada
Tipos 2 (Neuropática aguda) y Tipo 3 (Neuropática crónica): Formas Neuropáticas. 5% de los casos
- Todas las características del tipo 1
- Compromiso del SNC
- Mortalidad precoz:
- Tipo 2: Inicio antes de los 2 años. Expectativa de vida: 4 años
- Tipo 3: Inicio luego de los 2 años. Expectativa de vida: 40 años aprox.
- Mutación L444P es la más comúnmente asociada
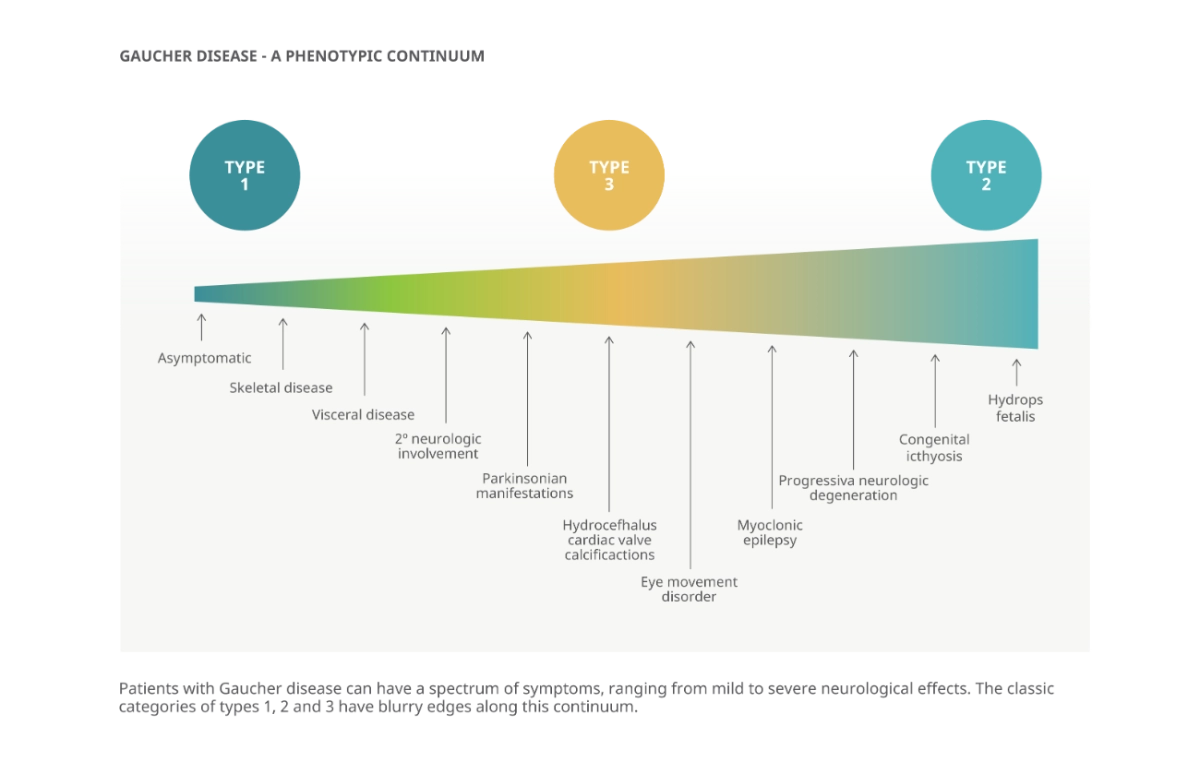
Clasificación fenotípica de la enfermedad de Gaucher
Diagnóstico de la enfermedad de Gaucher
1. Dosaje enzimático en DBS
Ante la sospecha de Enfermedad de Gaucher, se solicita un test de screening: el dosaje de actividad enzimática
(beta glucosidasa acida) en gotas de sangre en papel de filtro (DBS):
Ventajas:
- Simplicidad en el envío y transporte de las muestras: correo a temperatura ambiente
- Facilidad para la toma de muestra
- Reducción del tiempo de procesamiento y los volúmenes de reacción
- Estudios a gran escala: Screening
- Almacenamiento simple (hasta varios años).
Ante un resultado patológico de medición de actividad enzimática en DBS, se solicita la confirmación de diagnóstico con medición de actividad enzimática en leucocitos.
2. Dosaje enzimático en leucocitos
Muestra en sangre entera con heparina
Diagnóstico
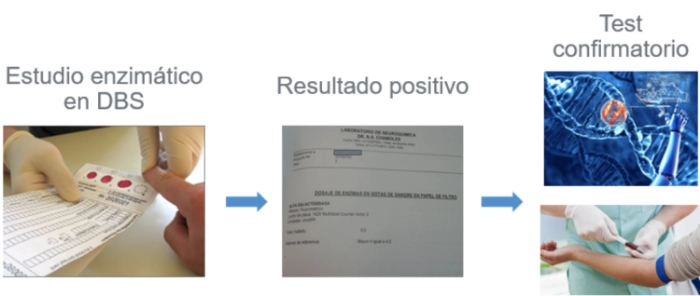
3. Estudio genético
La Enfermedad de Gaucher (EG) es una enfermedad autosómica recesiva causada por mutaciones bi-alélicas en el gen GBA.
El gen GBA comprende 11 exones y 10 intrones, que abarcan una secuencia de 7,6 kb y codifica una proteína madura de 497 aa.
Existe un pseudogen altamente homólogo (GBAP1) con la misma organización de exones e intrones que el gen funcionalLa genotipificación de los pacientes con EG puede ser un gran desafío, fundamental que el estudio genético lo realice un laboratorio con experiencia en EG.
EG es una enfermedad compleja en su heterogeneidad clínica, con una correlación limitada de genotipo/fenotipo.
Hasta la fecha, se han reportado más de 400 mutaciones en GBA causales de EG.
N370S y L444P son globalmente las mutaciones más prevalentes.
N370S se describe como "neuroprotectora": la presencia en un solo alelo en un paciente es predictivo de Gaucher tipo 1.
La homocigosidad para L444P generalmente se asocia con un EG tipo 3, y L444P en combinación con un alelo recombinante, generalmente se asocia con EG tipo 2.
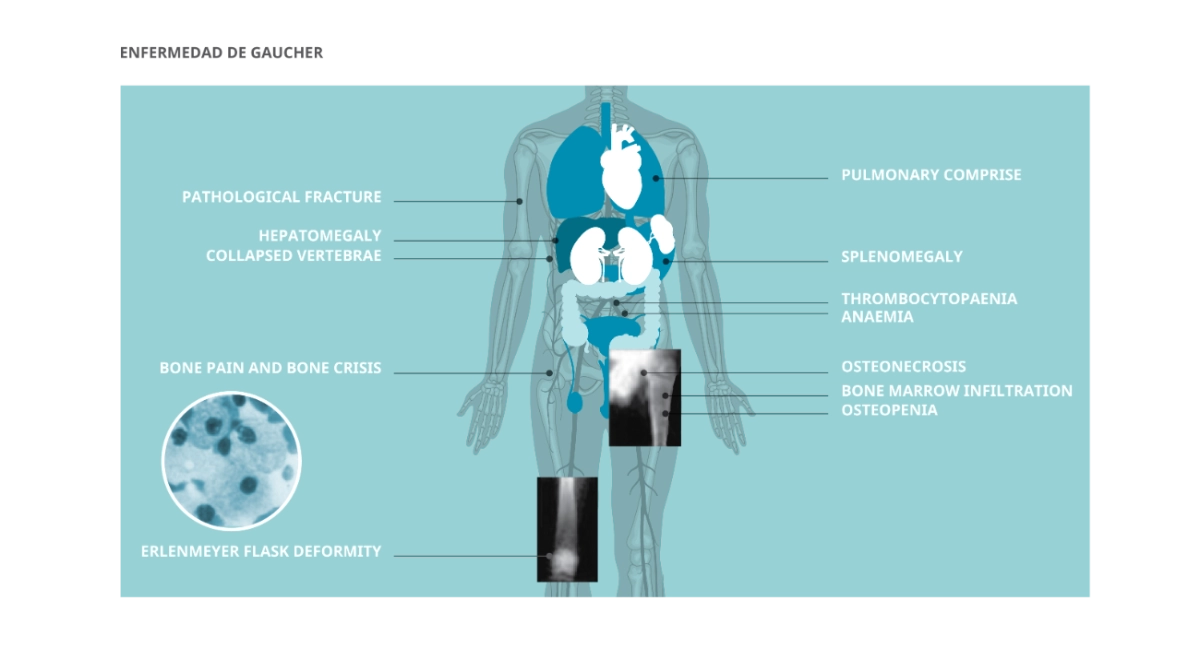
Debe sospecharse ante la combinación de hepatoesplenomegalia, anemia, trombocitopenia y compromiso óseo (dolor, fracturas, deformidad de Erlenmeyer en radiografía). La sospecha debe considerarse también ante esplenomegalia aislada de causa no determinada, citopenias crónicas sin explicación hematológica o necrosis ósea avascular en pacientes jóvenes. El diagnóstico promedia 3–4 años desde los síntomas iniciales, con consulta a 3 especialistas en promedio antes de confirmarse. Los errores diagnósticos más frecuentes son anemia crónica, enfermedad hemato-oncológica, cirrosis y esplenomegalia idiopática.
El diagnóstico se confirma mediante la medición de actividad de glucocerebrosidasa (GCasa) en leucocitos circulantes o cultivo de fibroblastos. El test de gota de sangre seca en papel de filtro (DBS) es útil para tamizaje orientativo pero requiere confirmación. El estudio molecular del gen GBA1 documenta la mutación específica y permite correlación genotipo-fenotipo y consejo genético familiar. En Argentina, el DIEL del Hospital de Clínicas realiza estos estudios en forma gratuita para pacientes de todo el país.
La enfermedad de Gaucher tipo 1 es la forma más frecuente (sin compromiso neurológico primario), con manifestaciones principalmente hematológicas, viscerales y óseas. La prevalencia es 1:40.000–60.000 en la población general y 1:800 en población judía asquenazí. El tipo 2 es una forma neuronopática aguda infantil con evolución fatal. El tipo 3 es una forma neuronopática crónica de inicio en la infancia o adolescencia. El conocimiento del tipo guía la selección terapéutica y el pronóstico.
Debes ser un profesional de la salud para continuar leyendo este contenido.
¿Eres un profesional de la salud?
Regístrate para obtener acceso exclusivo a las últimas noticias sobre avances científicos y a recursos que mejoren la vida de tus pacientes.
Login
Obtenga acceso a información para profesionales de la salud y edite los temas relevantes para usted en su perfil.
HCP Verification Required
REENVÍO ÉXITO
Por favor revise su correo electrónico para ver el correo electrónico de verificación.
Contenido restringido
No tienes el rol verificado correcto para ver este contenido.
Revisa tu bandeja de entrada de correo electrónico para activar tu cuenta
Se ha enviado un correo electrónico de verificación al correo electrónico con el que se registró. Para activar su cuenta, haga clic en el enlace de este correo electrónico. Si no ha recibido el correo electrónico de verificación, haga clic en el botón a continuación.
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.