
La enfermedad de Pompe es una enfermedad neuromuscular genética progresiva que puede afectar a pacientes de todas las edades1-3
La enfermedad de Pompe puede causar daño muscular que puede dar como resultado insuficiencia respiratoria, debilidad muscular progresiva con pérdida de la función motora, insuficiencia cardíaca y muerte prematura.1,2,4,5
Tipos de enfermedad de Pompe
Temas cubiertos en esta sección:
La enfermedad de Pompe es heterogénea, con una variedad de síntomas, edades de inicio y velocidad de progresión1,6
Los pacientes con enfermedad de Pompe presentan con mayor frecuencia signos de deterioro motor y respiratorio1,4,5

Aprenda a reconocer los signos y síntomas de la enfermedad de Pompe
Aproximadamente 1 de cada 40 000 personas en todo el mundo tiene la enfermedad de Pompe.7
Manifestaciones comunes
Aprenda a reconocer los signos y síntomas de la enfermedad de Pompe
El deterioro continuo de la función respiratoria es inevitable sin tratamiento y requerirá un manejo activo y precoz de la enfermedad.1,6,9-11

Origen y genética
En la enfermedad de Pompe, la actividad deficiente de la enzima α-glucosidasa ácida (GAA) conduce a un daño muscular progresivo y debilitante1-3
Origen
La enfermedad de Pompe fue descripta por primera vez en 1932 por el patólogo holandés Johannes C. Pompe, quien informó el caso de un bebé de 7 meses que murió por hipertrofia cardíaca idiopática. Se descubrió que este bebé tenía una acumulación masiva de glucógeno en muchos tejidos, pero predominantemente en los músculos esquelético y cardíaco. La enfermedad de Pompe puede afectar tanto a pacientes pediátricos como adultos.12,13
Deficiencia
En 1963, la enfermedad de Pompe se relacionó con una deficiencia hereditaria de la enzima lisosomal, GAA, responsable de la degradación del glucógeno en glucosa.12 La enfermedad de Pompe es una enfermedad genética autosómica recesiva.1 Las mutaciones heredadas en el gen que codifica la producción de GAA-gen 17q25.2 - q25.3, en el cromosoma 17- provocan una marcada deficiencia o ausencia de actividad de la enzima GAA en pacientes con enfermedad de Pompe.12,14
Destrucción
En la enfermedad de Pompe, la acumulación continua de glucógeno debido a la actividad deficiente de la enzima GAA hace que los lisosomas se hinchen y se rompan, lo que da como resultado daño celular. Esto, a su vez, conduce a una degeneración progresiva de los músculos esqueléticos y respiratorios (junto con el músculo cardíaco, principalmente en los bebés), lo que genera una pérdida de función.12,15
Acumulación de glucógeno en las células de un paciente con enfermedad de Pompe
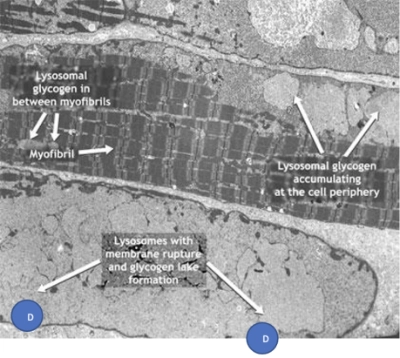
A. Miofibrillas
B. Glucógeno lisosomal entre miofibrillas
C. Glucógeno lisosomal que se acumula en la periferia de la célula
D. Lisosomas con ruptura de membrana y formación de lagunas de glucógeno
En pacientes con enfermedad de Pompe, la acumulación de glucógeno y el daño muscular resultante a menudo ocurren antes que cualquier signo o síntoma clínicamente detectable.
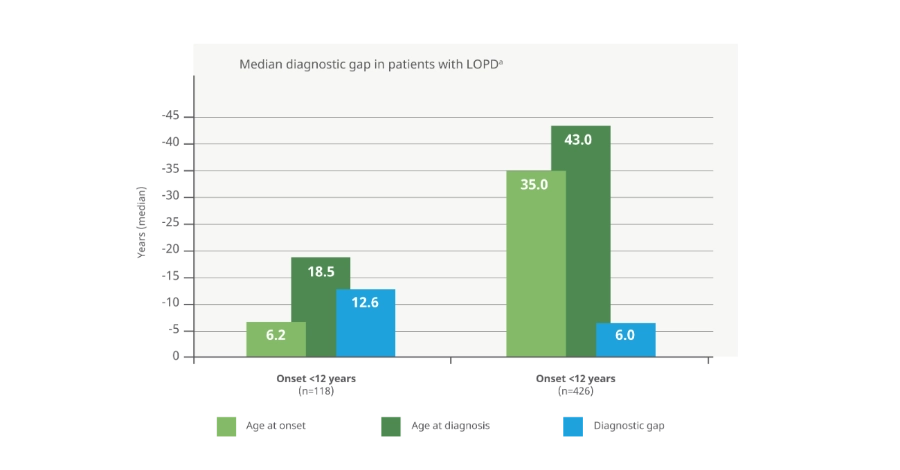
Retraso en el diagnóstico
Los pacientes con LOPD pueden experimentar una demora de entre 6 a 13 años antes de recibir un diagnóstico definitivo18
Sin un manejo adecuado, LOPD puede provocar un daño muscular progresivo y potencialmente irreversible, deterioro de la función respiratoria y disminución de la movilidad.1,2,19,20
Diagnostique y controle la enfermedad de Pompe de manera temprana para ayudar a reducir el impacto de la enfermedad.19
La acumulación progresiva de glucógeno y consecuentemente el daño muscular progresivo indican la necesidad de un diagnóstico precoz, inicio temprano de terapia y un activo manejo de los pacientes con enfermedad de Pompe.19
Enfermedad de Pompe: descripción
Temas desarrollados en esta sección:

Epidemiología de la enfermedad de Pompe
Incidencia, prevalencia y distribución étnica
Aproximadamente 1 de cada 40,000 personas nace con la enfermedad de Pompe1
Como ocurre con cualquier enfermedad poco frecuente, es difícil saber exactamente cuántas personas se ven realmente afectadas por la enfermedad de Pompe. Extrapolando las cifras de incidencia asumidas, se calcula que la prevalencia mundial actual puede ser de 5.000 a 10.000 personas.2,3
La incidencia de la enfermedad de Pompe puede variar entre poblaciones4
Varios estudios sugieren que la incidencia de la enfermedad de Pompe puede oscilar entre 1 en 40.000 y 1 en 300.000, dependiendo de la ubicación geográfica o el grupo étnico examinado.5
En los Países Bajos, existe una mayor incidencia de la forma tardía de la Enfermedad de Pompe (LOPD). Además, algunas mutaciones del gen GAA son más comunes en ciertas poblaciones. La forma infantil de la enfermedad de Pompe (IOPD) parece ser más frecuente entre las poblaciones afroamericana y china.5
¿Podría la enfermedad de Pompe pasar inadvertida en su consultorio?
Aprenda a identificar mejor a los pacientes que pueden estar viviendo con la enfermedad de Pompe.
Como trastorno autosómico recesivo, solo ocurre cuando un individuo hereda 2 alelos mutantes, 1 de cada progenitor. La mayoría de los pacientes son heterocigotos compuestos, lo que significa que han heredado 2 mutaciones diferentes.6-8
La enfermedad de Pompe es una afección hereditaria causada por mutaciones del gen α glucosidasa ácida (GAA) 17q25.2-q25.3, en el cromosoma 17.5,9
En general, la correlación genotipo-fenotipo no es muy bien entendida y puede existir una heterogeneidad clínica significativa entre pacientes con mutaciones similares o idénticas. Una excepción es la presencia de 2 mutaciones sin sentido, lo que deriva en una ausencia completa de actividad de la enzima GAA. Este genotipo da como resultado una presentación muy temprana de la enfermedad durante la infancia y una progresión rápida y grave. Sin embargo, se necesitan más estudios para comprender mejor las relaciones genotipo-fenotipo.
Si bien el análisis de las mutaciones no predice necesariamente el impacto de la enfermedad, es una herramienta importante que puede ayudar a confirmar un diagnóstico inicial y asistir en las pruebas familiares, de portadores y diagnósticos prenatales.13,14
Hasta la fecha, se han identificado más de 500 variantes de GAA18
No todas las variantes de GAA se consideran patógenas; siguen reportándose nuevas variantes y el Centro Pompe de la Universidad Erasmus de Rotterdam, Países Bajos, mantiene un catálogo actualizado.18
Fisiopatología de la enfermedad de Pompe
En la enfermedad de Pompe, la actividad deficiente de la enzima GAA, causada por una mutación genética subyacente, conduce a un daño muscular progresivo y debilitante12,13,16
Tanto en la enfermedad de Pompe de inicio infantil (IOPD) como en la de inicio tardío (LOPD), la actividad deficiente de la enzima GAA da como resultado una acumulación progresiva de glucógeno lisosomal en los miocitos de todo el cuerpo.5,12
La acumulación progresiva y anárquica de glucógeno causa que los lisosomas se hinchen y se rompan, lo que da como resultado17
- Daño celular irreversible17,18
- Destrucción progresiva del músculo esquelético (incluida la musculatura respiratoria) y músculo liso17,19
- Manifestaciones respiratorias, motoras, musculoesqueléticas, cardíacas, bulbares y gastrointestinales (GI) debilitantes12,19-22

Músculo funcional23
Los síntomas pueden no ser clínicamente detectables
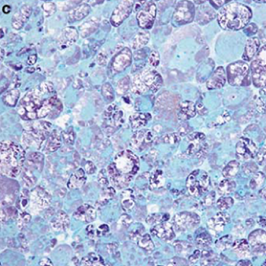
Músculo dañado23,24
Daño extenso del tejido muscular y función deteriorada
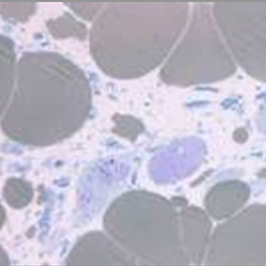
Músculo disfuncional a dañado23,24
Glucógeno citoplasmático con daño muscular leve a severo y disolución de fibrillas
Variabilidad de niveles enzimáticos
Si bien la enfermedad de Pompe siempre se describe por una actividad de GAA más baja de lo normal, el grado exacto de actividad enzimática residual varía entre los pacientes16:
- Los bebés con IOPD suelen tener menos del 1 % de la media normal de actividad de GAA16
- En niños y adultos con LOPD, la actividad de la enzima GAA suele estar entre el 1 % y el 40 % de lo normal16
En general, existe poca correlación entre la actividad de GAA residual y las manifestaciones clínicas en niños y adultos, aunque los bebés siempre estarán gravemente afectados.6,12,16,25
El Diagnostico y manejo temprano de enfermedad de Pompe ayudará a reducir la carga de la enfermedad26
La acumulación progresiva de glucógeno y el daño muscular continuo requieren un diagnóstico precoz y un tratamiento activo de los pacientes con enfermedad de Pompe.26
Cuadro clínico y evolución
Tópicos cubiertos en esta sección:
Manifestaciones comunes de la enfermedad de Pompe de inicio tardío (LOPD)
Aunque las manifestaciones de la enfermedad de Pompe son multisistémicas, los pacientes con LOPD suelen presentar signos de deterioro motor y respiratorio1-3
El compromiso respiratorio es un signo importante de daño muscular diafragmático en la enfermedad de Pompe que a menudo se ve eclipsado por pérdida de movilidad.4
Signos de deterioro de los músculos motores y respiratorios
.2023-02-27-09-02-11.png0/jcr:content.png)
- Debilidad en las piernas
- Dificultad para subir escaleras
- Dificultad para levantarse de una silla
- Caminar balanceando las caderas o rengueando
- Caídas frecuentes y problemas para correr o hacer deporte

- Disnea al hacer esfuerzo, p. ej. subir escaleras
- Dificultad para respirar, especialmente en decúbito dorsal
- Enfermedad pulmonar restrictiva y tos inefectiva
- Dificultad respiratoria nocturna, manifestada por apnea del sueño, dolores de cabeza matutinos o niveles elevados de CO2
- Hipoventilación durante el sueño
- Fatiga excesiva durante el día
- Intolerancia al ejercicio
LOPD en movimiento
Los pacientes que padecen LOPD pueden experimentar desafíos en una variedad de tareas diarias, desde subir escaleras hasta levantarse de la silla o alcanzar algo en altura. Utilizando la tecnología de captura de movimiento, los siguientes videos animados muestran a pacientes con distintos niveles de gravedad de la enfermedad realizando estas tareas diarias.
Manifestaciones de LOPD e IOPD
LOPD
La LOPD se puede diagnosticar tanto en niños como en adultos. La LOPD generalmente progresa a un ritmo más lento que en los pacientes con IOPD. A diferencia de la IOPD, los pacientes con LOPD tienen poca o ninguna afectación cardíaca. Sin embargo, la enfermedad de Pompe es implacablemente progresiva y se asocia con una morbilidad significativa o mortalidad prematura.
Los pacientes con LOPD presentan una gran heterogeneidad. Las manifestaciones clínicas más frecuentes son la debilidad progresiva de los músculos proximales y la insuficiencia respiratoria que, en última instancia, conducen a la pérdida de la deambulación y a la necesidad de asistencia respiratoria.
Manifestaciones de la enfermedad en pacientes con LOPD
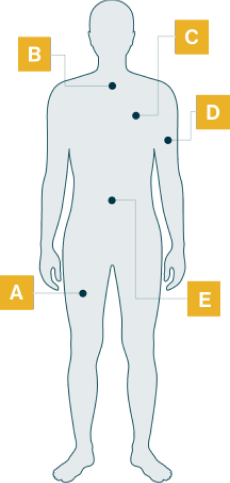

- Dificultad para elevar los brazos
- Debilidad en las piernas
- Dificultad para subir escaleras
- Dificultad para levantarse de una silla
- Caminar balanceando las caderas, cojera
- Caídas frecuentes
- Dificultad para correr y hacer deportes
- Dificultad para hablar
- Dificultad para tragar
- Debilidad de los músculos faciales (p. ej., ptosis)
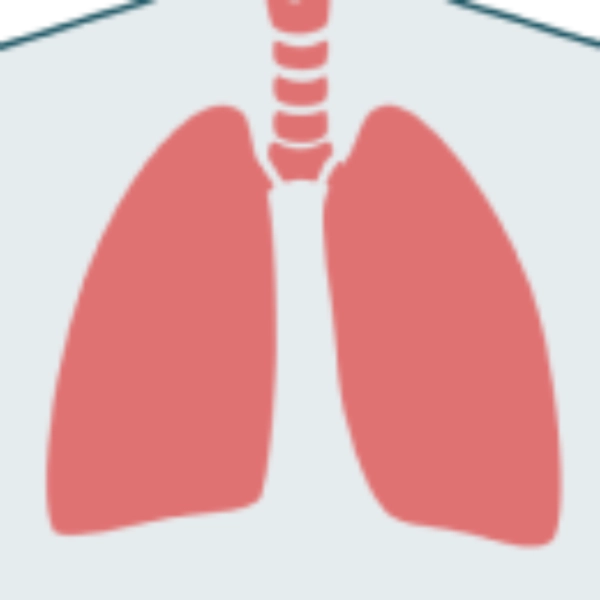
- Disnea al hacer esfuerzo, p. ej. subir escaleras
- Dificultad para respirar, especialmente en decúbito dorsal
- Enfermedad pulmonar restrictiva y tos alterada
- Dificultad respiratoria nocturna, indicada por apnea del sueño, dolores de cabeza matutinos o niveles elevados de dióxido de carbono
- Hipoventilación durante el sueño
- Fatiga excesiva durante el día
- Intolerancia al ejercicio

- Escoliosis
- Osteoporosis
- Osteopenia
- Fracturas vertebrales
- HiperCKemia persistente inexplicable (1,5 a 15 veces los niveles normales, ~300-2000 U/L)
- Pérdida de peso inexplicable
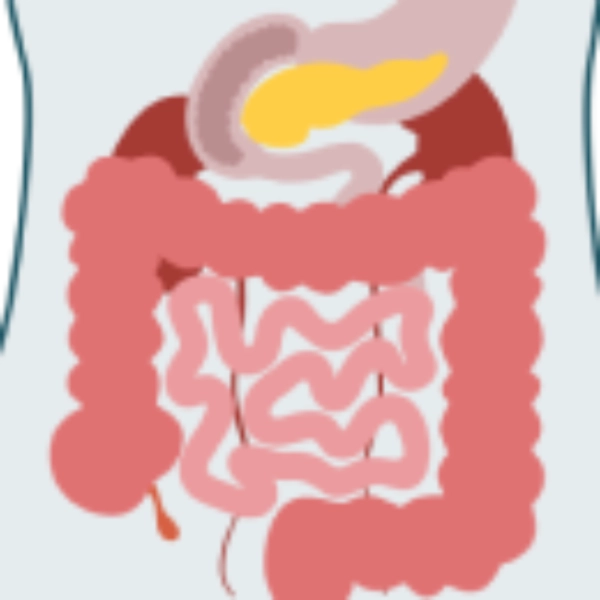
- Incontinencia imperiosa fecal/urinaria
- Síntomas de malestar gastrointestinal
IOPD
Los pacientes con IOPD suelen presentar hipotonía pronunciada y cardiomegalia grave. Otros signos incluyen infecciones respiratorias frecuentes, incapacidad para alcanzar los hitos motores y dificultad para alimentarse.
Manifestaciones de la enfermedad en pacientes con IOPD
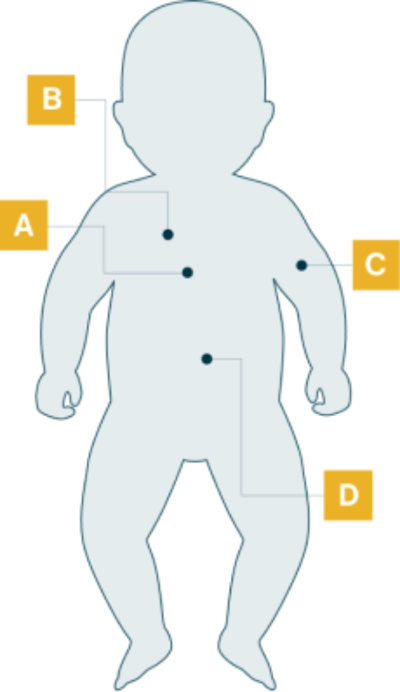
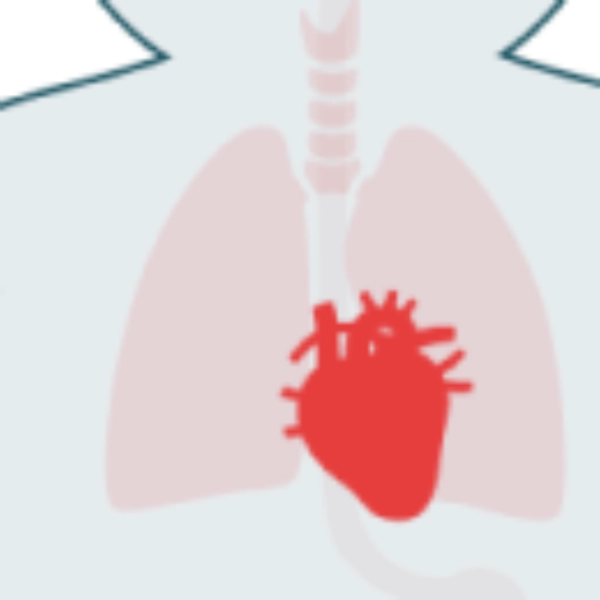
- Cardiomegalia
- Miocardiopatía progresiva
- Afectación progresiva de los músculos respiratorios
- Infecciones respiratorias frecuentes
- Respiración alterada por el sueño
.2023-10-09-14-47-08.png)
- Debilidad muscular progresiva
- Hipotonía profunda/ausencia de sostén cefálico/"bebé flácido"
- Hitos motores retrasados
.2023-10-09-14-48-40.png)
- Macroglosia
- Dificultades para la alimentación
- Retraso del crecimiento/poco aumento de peso
- Hepatomegalia
Realice una prueba temprana para detectar la enfermedad de Pompe si advierte estos signos o síntomas comunes en su consultorio
La acumulación progresiva de glucógeno y el daño muscular continuo pueden conducir a una multitud de manifestaciones debilitantes y potencialmente mortales
En pacientes con enfermedad de Pompe, el daño muscular continuo puede provocar insuficiencia respiratoria progresiva y una variedad de otros síntomas debilitantes y potencialmente mortales.
Pacientes con LOPD que experimentaron dificultades con las tareas en el momento del diagnóstico a, b
En un estudio observacional de 12 meses en pacientes con LOPD a los que se les administró la prueba de caminata de 6 minutos, hubo una diferencia de casi 150 metros en la resistencia funcional entre los siguientes grupos de pacientes:

Aquellos que requerían asistencia motora para caminar lograron una distancia promedio de 230,4 metros (37,9 % de lo normal previsto)

Aquellos que podían caminar sin ayuda alcanzaron una distancia promedio de 380,3 metros (59,5 % de lo normal previsto)
(Basado en un análisis retrospectivo del estado funcional y de salud documentado de 42 pacientes con LOPD (13 niños afectados [edad media en el momento del diagnóstico: 10 años; rango: 0-16 años] y 29 pacientes adultos [edad media en el momento del diagnóstico: 43 años; rango: 24-68 años]) durante un año después del diagnóstico.
(Según la evaluación de la prueba de función motora rápida (QMFT), una medida del grado en que los pacientes experimentan dificultades con 16 acciones diferentes (p. ej., incorporarse, saltar, etc.). Las puntuaciones por acción pueden variar de 0 (no es posible o no se ejecuta por otro motivo) a 4 (acción completada)
Realice una prueba temprana para detectar la enfermedad de Pompe si advierte estos signos o síntomas comunes en su consultorio
Insuficiencia respiratoria
En la LOPD, el deterioro respiratorio y la amenaza de insuficiencia respiratoria requieren un diagnóstico precoz y un tratamiento activo
Modelos de progresión del deterioro respiratorio, medida por la capacidad vital forzada (CVF), en pacientes con LOPD no tratados c
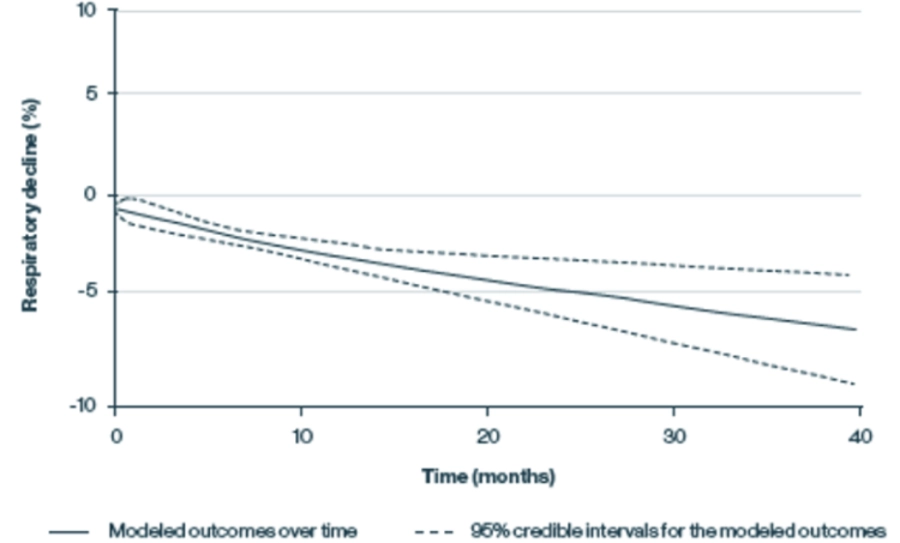
Adaptado de Schoser B et al, J Neurol, 2016
La función respiratoria promedio, medida por CVF, al ingresar al estudio fue del 61 %, lo que refleja una enfermedad pulmonar restrictiva moderada.
La función respiratoria disminuyó de manera constante, incluida una disminución del 2,3 % después de 12 meses y una disminución del 6,2 % después de 48 meses.
(Basado en un metaanálisis de 19 estudios de pacientes con LOPD. Los resultados continuos se modelaron mediante el metaanálisis de polinomios fraccionados, que estima el desarrollo de los resultados a lo largo del tiempo.
En la IOPD, la insuficiencia cardiorrespiratoria es la principal causa de muerte prematura
Los síntomas se presentan en la primera infancia, progresan rápidamente y, por lo general, conducen a la muerte alrededor de los 2 años.
.2023-10-09-15-21-55.png)
Estudio de caso: paciente con LOPD
Mujer de 39 años con insuficiencia respiratoria progresiva (Simulación de paciente - imagen de archivo)
En pacientes con enfermedad de Pompe, el daño muscular continuo puede provocar insuficiencia respiratoria progresiva y una variedad de otros síntomas debilitantes y potencialmente mortales.
Características clínicas
- Progresiva dificultad para respirar
- Dificultad para respirar al hacer esfuerzo o pocos minutos después de acostarse
- Disnea excesiva al nadar
- Hipersomnia diurna con dolores de cabeza matutinos asociados
Historial médico
- Puntuación de Epworth: 21 (de 24)
- Émbolos pulmonares no provocados 6 años antes
- Síndrome de congestión venosa pélvica
- Sometida a una terapia de anticoagulación persistente
- La escala de somnolencia de Epworth mide la somnolencia diurna a través de 8 preguntas en una escala de 4 puntos, de 0 ("nunca dormitaría") a 3 ("alta probabilidad de dormitar"), que evalúan la propensión de un paciente a dormitar o quedarse dormido durante actividades cotidianas comunes
Evaluación clínica
- Resultados de laboratorio
- Saturación de oxígeno en aire ambiente, 97
- Conteo sanguíneo completo: normal
- Proteína C reactiva, normal
- Creatina quinasa, levemente elevada (624 U/L)
- Evaluación de movilidad
- Andar levemente oscilante
- Debilidad limitada en la flexión de la cader
- Resultados de la prueba de función respiratoria
- CVF en posición sentada, 2,29 L; 82 % previsto
- CVF en posición supina, 0,92 L; 33 % previsto
- Evaluación diurna de gasometría arterial
- Presión parcial de dióxido de carbono, 54 mm Hg (límite superior de lo normal, 45 mm Hg)
- Presión parcial de oxígeno, 85 mm Hg (rango normal, 85 a 95 mm Hg)
- Polisomnografía nocturna
- Desaturación severa durante el sueño con movimientos oculares rápidos (nadir 68 % de aire ambiental)Hg)
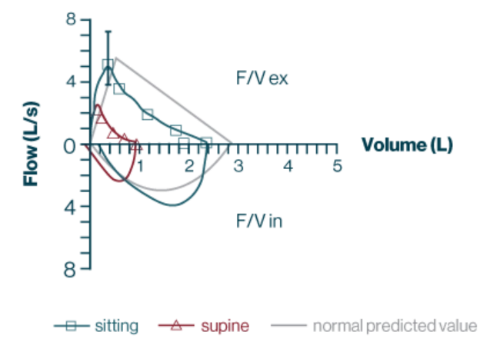
Abreviaturas: F/V es.: porción espiratoria del bucle de volumen de flujo; F/V in.: parte inspiratoria del bucle de volumen de flujo.
- Resultados de las pruebas de diagnóstico
- Prueba de gota de sangre: nivel de actividad de la enzima α-glucosidasa: < 0,1 (rango normal, 0,3-3,0)
- Intervenciones terapéuticas
- Ventilación binivel en modo temporizado-espontáneo
- Esperando el inicio de la terapia específica contra la enfermedad

La enfermedad de Pompe de inicio tardío se presenta típicamente con debilidad muscular proximal progresiva de predominio en miembros inferiores y musculatura axial, intolerancia al ejercicio, elevación de CK sérica, y compromiso respiratorio que puede manifestarse como disnea de esfuerzo, ortopnea o insuficiencia respiratoria, incluso antes del compromiso motor severo. La compromiso diafragmático precoz es característica distintiva respecto a otras miopatías. El diagnóstico se demora en promedio varios años desde los síntomas, con frecuente confusión inicial con distrofia muscular de cinturas u otras miopatías metabólicas.
Debe sospecharse en adultos con debilidad muscular proximal de causa no determinada, especialmente si se acompaña de compromiso respiratorio desproporcionado al déficit motor, elevación crónica de CK, síntomas nocturnos como cefalea matinal u ortopnea, o antecedentes familiares de miopatía. Las especialidades con mayor probabilidad de encontrar estos pacientes son neurología, neumonología, cardiología (en formas con cardiomiopatía), medicina del deporte y rehabilitación. La detección mediante tamizaje enzimático con DBS tiene alta sensibilidad y especificidad.
Según el Consenso Argentino para el Diagnóstico, Seguimiento y Tratamiento de la Enfermedad de Pompe (Neurología Argentina), el proceso diagnóstico comienza con la medición de actividad de GAA en gota de sangre seca en papel de filtro (DBS), desarrollada originalmente en Argentina por el Dr. Néstor Chamoles. Un resultado bajo debe confirmarse con medición enzimática en leucocitos o fibroblastos, seguida de estudio molecular del gen GAA para identificar las variantes patogénicas y confirmar el diagnóstico.
Debes ser un profesional de la salud para continuar leyendo este contenido.
¿Eres un profesional de la salud?
Regístrate para obtener acceso exclusivo a las últimas noticias sobre avances científicos y a recursos que mejoren la vida de tus pacientes.
Login
Obtenga acceso a información para profesionales de la salud y edite los temas relevantes para usted en su perfil.
HCP Verification Required
REENVÍO ÉXITO
Por favor revise su correo electrónico para ver el correo electrónico de verificación.
Contenido restringido
No tienes el rol verificado correcto para ver este contenido.
Revisa tu bandeja de entrada de correo electrónico para activar tu cuenta
Se ha enviado un correo electrónico de verificación al correo electrónico con el que se registró. Para activar su cuenta, haga clic en el enlace de este correo electrónico. Si no ha recibido el correo electrónico de verificación, haga clic en el botón a continuación.
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.