
Enfermedad de Fabry: sospecha clínica y diagnóstico para profesionales de la salud
Guía clínica orientada a profesionales de la salud sobre la sospecha y diagnóstico de la enfermedad de Fabry, un trastorno lisosomal ligado al cromosoma X causado por deficiencia de alfa-galactosidasa A. Incluye señales de alerta clínicas, biomarcadores, algoritmos diagnósticos y enfoque multidisciplinario actualizado según las recomendaciones argentinas 2025.
Sobre la enfermedad de Fabry
Descubriendo la enfermedad de Fabry
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosomal ligado al cromosoma X que afecta a hombres, mujeres y niños de todas las etnias.1,2
La enfermedad de Fabry es una enfermedad rara hereditaria y progresiva, que afecta a varios sistemas del cuerpo, incluidos el corazón, los riñones, el sistema nervioso y la piel. Esta condición es provocada por una mutación en el gen GLA, que interfiere con la producción de una enzima llamada alfa-galactosidasa A, la cual es esencial para descomponer ciertos lípidos en el organismo. La falta o insuficiencia de esta enzima provoca la acumulación de lípidos en las células, lo que conlleva a problemas que afectan múltiples sistemas.
No te pierdas estos enlaces clave.

Aviso de Farmacovigilancia
¿Qué es la enfermedad de Fabry?

La enfermedad de Fabry es multisistémica. Conoce más acerca de sus manifestaciones:
Generalidades de las Enfermedad de Fabry
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosomal ligado al cromosoma X que afecta a hombres, mujeres y niños de todas las etnias.1,2 Su prevalencia se estima entre 1: 40.000.1,3
Es un trastorno multisistémico progresivo que puede comprometer órganos vitales como los riñones, el corazón y el cerebro incrementando la morbimortalidad de los pacientes.2,4
Los primeros signos y síntomas de la enfermedad de Fabry pueden comenzar en la infancia o la adolescencia incluyendo dolor, trastornos gastrointestinales, angioqueratomas, hipohidrosis y proteinuria, entre otros. 5 A medida que progresa puede producir deterioro en la función renal, miocardiopatía hipertrófica y aumento del riesgo a desarrollar eventos graves tales como enfermedad renal terminal, accidente cerebrovascular, insuficiencia cardíaca, arritmias y muerte prematura.1,5
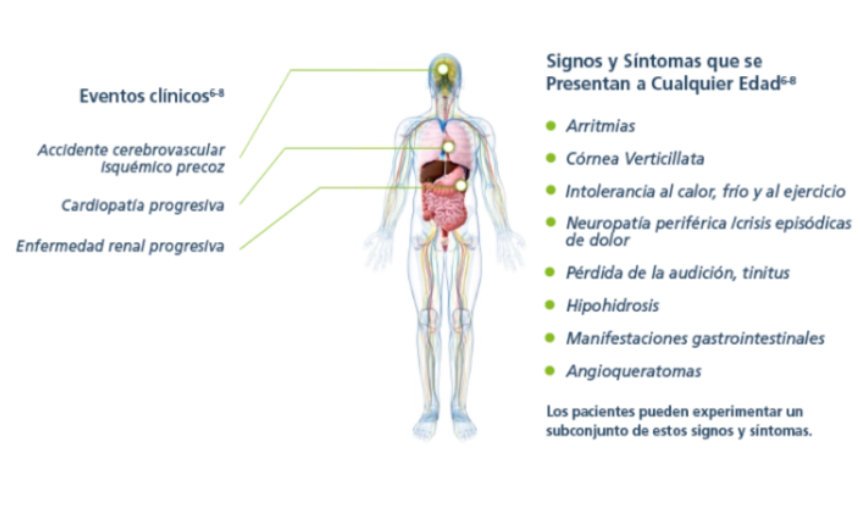
La enfermedad de Fabry es multisistémica. Conoce más acerca de sus manifestaciones:
Signos y síntomas por etapa de la vida
Los pacientes pueden experimentar algunos o todos estos síntomas.

Fisiopatología
En la enfermedad de Fabry, las mutaciones en el gen GLA, ubicado en el cromosoma X, producen defectos en la síntesis y/o función de la alfa-galactosidasa A (α- Gal A).9
La α- Gal A es una enzima lisosomal que metaboliza la globotriaosilceramida (también conocida como GL-3 o Gb3) y evita que se acumule.10
La deficiencia total o parcial de α- Gal A conduce a la acumulación progresiva de glicoesfingolípidos, particularmente GL-3, en los lisosomas de numerosos tipos celulares.5
En la enfermedad de Fabry, la acumulación de GL-3 comienza en el útero, y continúa durante toda la vida.1,2 Con el avance de la edad su acumulación, particularmente en el endotelio vascular, conduce a insuficiencia renal, enfermedad cardíaca, accidente cerebrovascular y muerte prematura, generalmente en la cuarta o quinta década de la vida.4,5,10,11
Las manifestaciones clínicas de la enfermedad de Fabry abarcan un amplio espectro en términos de severidad y generalmente están correlacionadas con el nivel de actividad residual de α- Gal A
En la enfermedad de Fabry, las mutaciones en el gen GLA, ubicado en el cromosoma X, producen defectos en la síntesis y/o función de la alfa-galactosidasa A (α- Gal A).9
La α- Gal A es una enzima lisosomal que metaboliza la globotriaosilceramida (también conocida como GL-3 o Gb3) y evita que se acumule.10
La deficiencia total o parcial de α- Gal A conduce a la acumulación progresiva de glicoesfingolípidos, particularmente GL-3, en los lisosomas de numerosos tipos celulares.5
En la enfermedad de Fabry, la acumulación de GL-3 comienza en el útero, y continúa durante toda la vida.1,2 Con el avance de la edad su acumulación, particularmente en el endotelio vascular, conduce a insuficiencia renal, enfermedad cardíaca, accidente cerebrovascular y muerte prematura, generalmente en la cuarta o quinta década de la vida.4,5,10,11
Las manifestaciones clínicas de la enfermedad de Fabry abarcan un amplio espectro en términos de severidad y generalmente están correlacionadas con el nivel de actividad residual de α- Gal A
La enfermedad de Fabry es un trastorno genético; en consecuencia la detección familiar es de vital importancia.
Por cada paciente diagnosticado con Fabry, hay una familia que está en riesgo
Por cada paciente índice diagnosticado, se puede identificar a un promedio de 5 miembros adicionales de la familia que están afectados.
Se han identificado más de 900 mutaciones en el gen GLA asociado con la enfermedad de Fabry.9,12 La mayoría de las familias tienen mutaciones específicas.2
Como es una enfermedad ligada al cromosoma X, los hombres y las mujeres pueden transmitir la mutación genética que causa la enfermedad de Fabry. Además es importante destacar que las mujeres no son simplemente portadoras.1,5,13 Los hombres tienen una probabilidad del 100% de transmitir el gen mutado a sus hijas y no a sus hijos varones, mientras que las mujeres con enfermedad de Fabry tienen una probabilidad del 50% de transmitir el gen mutado a cada hija e hijo.5
Las mujeres con el gen GLA mutado pueden desarrollar Fabry, pero las manifestaciones pueden ser heterogéneas debido a la inactivación aleatoria del cromosoma X específica de órganos y tejidos.
Fenotipos14-16
Se han descripto dos formas de presentación para la enfermedad de Fabry: el fenotipo clásico de afectación multisistémica (Enfermedad de Fabry tipo 1) y las formas de inicio tardío (Enfermedad de Fabry tipo 2) que suelen afectar un solo órgano blanco.

El camino al diagnóstico
En un estudio basado en el Registro de Fabry, los retrasos en el diagnóstico y el diagnóstico erróneo fueron comunes debido a la naturaleza inespecífica y heterogénea de los primeros síntomas de la enfermedad de Fabry.8

A menudo, la enfermedad de Fabry se diagnostica erróneamente y se confunde con artritis reumatoidea o artritis idiopática juvenil, fiebre reumática, síndrome de Raynaud, lupus eritematoso sistémico o esclerosis múltiple.2,5
Conectar los síntomas aparentemente no relacionados con la enfermedad de Fabry puede ayudar a evitar los retrasos diagnósticos e iniciar la terapia a tiempo.10
La importancia de la detección de la enfermedad de Fabry en las poblaciones de alto riesgo
Aunque a la enfermedad de Fabry se la considera una enfermedad poco frecuente, la prevalencia en los pacientes con ciertos trastornos como enfermedad renal crónica inexplicada, miocardiopatía hipertrófica, y accidente cerebrovascular prematuro, es mayor que en la población general.17-19 Por consiguiente, es importante considerar la EF dentro de los diagnósticos diferenciales.
Las mujeres con enfermedad de Fabry
Según el Registro, el 69,4% de las mujeres con la enfermedad de Fabry desarrollan síntomas relacionados con la afección con el tiempo.8
Se sabe que las mujeres con EF presentan un curso clínico heterogéneo, por lo que generalmente sus síntomas se desarrollan en formas más tardía a diferencia de los hombres.13 Este rango de heterogeneidad en la sintomatología hace que el diagnóstico de Fabry en mujeres sea especialmente desafiante.
En las mujeres, esta variabilidad en la presentación de la EF se debe a los patrones de inactivación aleatorios del cromosoma X. Cada órgano en el cuerpo de una mujer tiene su propio patrón de inactivación aleatoria y conduce a variaciones en la expresión de la enfermedad.13
La actividad enzimática en las mujeres puede estar levemente disminuida o en rangos normales. En un estudio transversal de 57 mujeres heterocigotas sintomáticas, algunas con actividad enzimática normal presentaban anomalías cardíacas, renales o cerebrovasculares.22
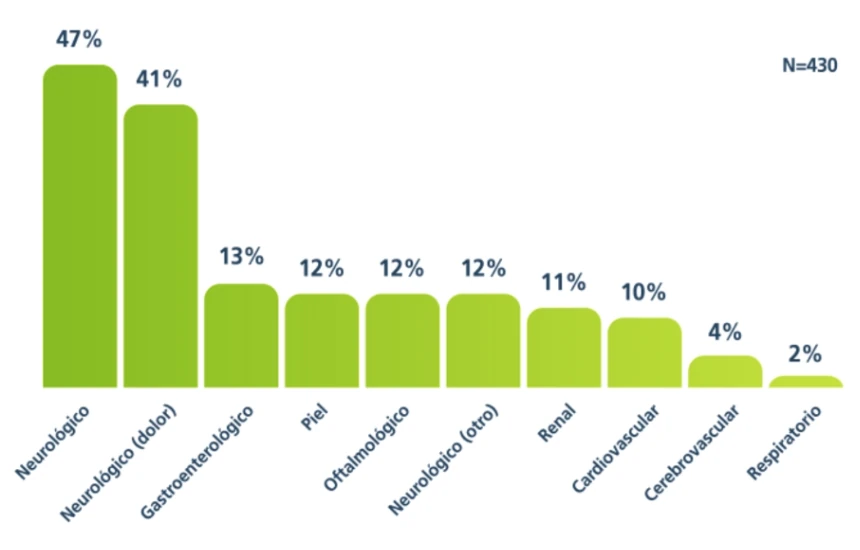
La enfermedad de Fabry en tu consulta
El paciente Fabry puede presentarse a la consulta médica de múltiples especialidades debido al compromiso multisistémico que caracteriza a esta enfermedad.
La enfermedad renal es una complicación importante de la enfermedad de Fabry.10,20 La prevalencia de la EF en la población en diálisis es aproximadamente entre 100 a 1000 veces mayor que en la población general.17-19,24,25 Los pacientes con enfermedad de Fabry tienen un alto riesgo de progresar a enfermedad renal con requerimiento de diálisis a una edad temprana (30 a 50 años, y a veces en la adolescencia).23 La prevalencia de la enfermedad renal crónica en los pacientes con enfermedad de Fabry es aproximadamente 5 veces mayor que en la población general.8,26
El daño renal como resultado de la acumulación de GL-3 en varias células renales puede iniciarse ya en primera década de la vida, a menudo precediendo las alteraciones de laboratorio y el inicio de síntomas.23,27
En hombres y mujeres, la acumulación progresiva de GL-3 en los podocitos, se manifiesta más tarde como proteinuria y reducción de la tasa de filtrado glomerular reducida con progresión en el tiempo a una enfermedad renal crónica.28,29
La inclusion de la enfermedad de Fabry en el diagnostico diferencial de la enfermedad renal crónica no explicada es clave para identificar a los pacientes y sus familias.
La European Renal Best Practice y las Kidney Disease Improving Global Outcomes (KDIGO) recomiendan la detección de hombres menores de 50 años y mujeres de cualquier edad con ERC de causa desconocida y otros síntomas asociados con la enfermedad de Fabry.31
Si sospechas que un paciente puede tener la enfermedad de Fabry, considera hacerle dosaje de la actividad enzimática en sangre seca (si es hombre) y dosaje del lyso GL-3 (si es mujer).
La enfermedad cardiaca es la causa mas comun de muerte en pacientes con enfermedad de Fabry.27 Se debe considerar esta enfermedad ante manifestaciones cardíacas de causa desconocida. La prevalencia de la enfermedad de Fabry en pacientes con hipertrofia ventricular izquierda (HVI) o miocardiopatía hipertrófica (MCH) se estima en al menos 1 de cada 100.32-34
Se detecto que el 49% de los hombres y el 35% de las mujeres con la enfermedad de Fabry presentaron un evento cardíaco a una edad promedio de 36 y 44 años, respectivamente, y los eventos cardíacos pueden aparecer tan precozmente como en la adolescencia.30
La hipertrofia cardíaca, la fibrosis y las anomalias en la conduccion pueden ser causadas por la acumulacion de GL-3 en los cardiomiocitos, lo que afecta negativamente la estructura y la funcion cardíaca. Otras manifestaciones cardíacas pueden incluir: Trastornos en el ECG como: intervalos PR cortos, bloqueo AV, anomalías de repolarización, cambios ST-T y por otro lado arritmias.35 La acumulación persistente de GL-3 en los cardiomiocitos a traves del tiempo puede conducir a HVI, lo que finalmente resulta en insuficiencia cardíaca.36
Incluir la enfermedad de Fabry en el diagnóstico diferencial de HVI o MCH con causa desconocida es clave para identificar pacientes y familias.
Las recomendaciones de la Sociedad Europea de Cardiología promueven una búsqueda sistemática de la causa subyacente de la HVI, incluidas las pruebas de laboratorio especializadas y el análisis genético.26 La detección de pacientes con HVI o MCH con causa desconocida para la enfermedad de Fabry es clave para identificar a los pacientes y sus familias.
Si sospechas que un paciente puede tener la enfermedad de Fabry, considera hacerle dosaje de la actividad enzimática en sangre seca (si es hombre) y dosaje del lyso GL-3 (si es mujer).
Las estimaciones de prevalencia de la enfermedad de Fabry en pacientes con accidente cerebrovascular prematuro (en menores de 45 años) varían del 1% a casi el 5% más que en la población general.42
Según los datos del registro, la edad media del primer accidente cerebrovascular es de 39 años en hombres y 46 años en mujeres.5
En la enfermedad de Fabry, el GL-3 puede acumularse en las neuronas periféricas y centrales, así como en los vasos sanguíneos cerebrales, causando:42,43
- Dolor neuropático y síntomas autonómicos, debido al daño a las neuronas y axones periféricos.
- Deterioro cognitivo y trastornos del estado de ánimo, como lo demuestran las lesiones de la sustancia blanca.
SNP (sistema nervioso periférico): Dolor neuropático
La acumulación de GL-3 causa daño neuronal que afecta principalmente a las fibras nerviosas pequeñas de los sistemas nerviosos somáticos y autónomos periféricos. El dolor es uno de los primeros síntomas de Fabry, presente en el 60% al 80% de los niños y niñas clásicamente afectados, lo que tiene un efecto en su calidad de vida. Los niños generalmente tienen una edad más temprana de inicio de síntomas que las niñas. Se han descrito dos tipos de dolor:5
- Crisis episódicas ("crisis Fabry"): dolor agonizante que se origina en las extremidades y se irradia hacia adentro de las extremidades y otras partes del cuerpo
- Dolor crónico: parestesias con ardor y hormigueo
SNC (sistema nervioso central)
Los primeros signos neuropáticos periféricos de la enfermedad de Fabry suelen ir seguidos de complicaciones cerebrovasculares y/o disfunción autonómica en la edad adulta.5
La enfermedad de Fabry a menudo se diagnostica erróneamente como esclerosis múltiple (EM) debido a las lesiones en la sustancia blanca.42,44
Algunas de las características neurológicas más devastadoras de la enfermedad de Fabry son causadas por las lesiones cerebrovasculares que son el resultado del compromiso multifocal de pequeños vasos sanguíneos. La afectación cerebrovascular puede conducir a una amplia variedad de signos y síntomas que varían de leves a graves, incluyendo cefalea, vértigo/mareos, accidentes isquémicos transitorios y accidentes cerebrovasculares.
Si sospechás que un paciente puede tener la enfermedad de Fabry, considerá hacerle dosaje de la actividad enzimática en sangre seca (si es hombre) y/o dosaje del lyso GL-3 (si es mujer).
Uno de los signos más precoces y frecuentes de la enfermedad de Fabry es la córnea verticillata, que se observa en el 80% de los pacientes con enfermedad de Fabry. Se puede detectar con un examen de rutina con lámpara de hendidura.50
Córnea verticillata distintiva en Fabry 2,34,50
Los depósitos corneales presentan un patrón simétrico, bilateral, en forma de espiral de depósitos epiteliales corneales GL-3 polvorientos, blancos, amarillos o de color café que emanan de un solo vórtice.50
Una serie de medicamentos, como la amiodarona y la cloroquina, también pueden causar este fenómeno.
Además de las opacidades corneales, los pacientes con enfermedad de Fabry pueden presentar⁵⁰:
- Cataratas capsulares posteriores con depósitos blanquecinos de material granular (catarata de Fabry).
- Dilatación aneurismática de las vénulas de paredes delgadas en la conjuntiva bulbar.
- Tortuosidad de leve a marcada y angulación del vaso retiniano.
La característica clínica más reconocible de la enfermedad de Fabry son los angioqueratomas, que tienen las siguientes características1,2:
- Lesiones cutáneas de color rojo oscuro o púrpura.
- Rango de tamaño desde una cabeza de alfiler hasta varios milímetros de diámetro.
- Vitropresión negativa.
- Distribución en traje de baño nalgas, ingle, periumbilical y parte superior de los muslos.
Angioqueratomas en la enfermedad de Fabry1,2
Angiectasias características de color rojo oscuro a negro azulado que se encuentran típicamente entre las regiones del muslo (izquierda) y el ombligo (derecha) ("distribución en traje de baño"). Usado con permiso del Dr. RJ. Desnick, PhD.
Las lesiones generalmente aparecen en la adolescencia o en la edad adulta y progresan con la edad.2 Los angioqueratomas son casi universales en hemizigotos masculinos clásicos; ocurren aproximadamente en el 30% de las mujeres heterocigotas clásicas.1
La acumulación de GL-3 puede comenzar en el útero y continuar durante toda la vida. En su forma clásica el inicio clínico de la enfermedad de Fabry ocurre en la infancia y su presentación puede no ser fácilmente reconocida. Los signos y síntomas a menudo no son tomados en cuenta como causa de enfermedad crónica o se atribuyen erróneamente a otros trastornos (artritis idiopática juvenil, fiebre reumática, síndrome de Raynaud, polineuropatía desmielinizante inflamatoria crónica y lupus eritematoso sistémico).
El curso clínico temprano de la enfermedad de Fabry generalmente involucra signos y síntomas que afectan principalmente la calidad de vida: dolor crónico, angioqueratomas, hipohidrosis, intolerancia al calor y al frío, y síntomas gastrointestinales.2,22
El dolor es uno de los primeros síntomas, presentándose en el 60% al 80% de los niños y niñas afectados por la variante clásica (tipo 1), lo que afecta su calidad de vida. Los niños comienzan con sintomatología más tempranamente que las niñas.5
El compromiso gastrointestinal (dolor abdominal postprandial, diarrea, náuseas y vómitos) suele pasar desapercibido siendo causa frecuente de mal progreso ponderal en la infancia. El síndrome del intestino irritable (SII) es un diagnóstico diferencial.5
En un estudio pediátrico basado en el Registro de Fabry, los niños y niñas con fenotipo clásico ( tipo 1) comienzan a desarrollar síntomas a una edad temprana (edad media de 6 años para niños [n = 194] y niñas [n=158]).46,51
La acumulación de GL-3 puede comenzar en el útero y continuar durante toda la vida. En su forma clásica el inicio clínico de la enfermedad de Fabry ocurre en la infancia y su presentación puede no ser fácilmente reconocida. Los signos y síntomas a menudo no son tomados en cuenta como causa de enfermedad crónica o se atribuyen erróneamente a otros trastornos (artritis idiopática juvenil, fiebre reumática, síndrome de Raynaud, polineuropatía desmielinizante inflamatoria crónica y lupus eritematoso sistémico).
El curso clínico temprano de la enfermedad de Fabry generalmente involucra signos y síntomas que afectan principalmente la calidad de vida: dolor crónico, angioqueratomas, hipohidrosis, intolerancia al calor y al frío, y síntomas gastrointestinales.2,22
El dolor es uno de los primeros síntomas, presentándose en el 60% al 80% de los niños y niñas afectados por la variante clásica (tipo 1), lo que afecta su calidad de vida. Los niños comienzan con sintomatología más tempranamente que las niñas.5
El compromiso gastrointestinal (dolor abdominal postprandial, diarrea, náuseas y vómitos) suele pasar desapercibido siendo causa frecuente de mal progreso ponderal en la infancia. El síndrome del intestino irritable (SII) es un diagnóstico diferencial.5
En un estudio pediátrico basado en el Registro de Fabry, los niños y niñas con fenotipo clásico ( tipo 1) comienzan a desarrollar síntomas a una edad temprana (edad media de 6 años para niños [n = 194] y niñas [n=158]).46,51
Los síntomas gastrointestinales (Gl) de la enfermedad de Fabry pueden estar relacionados con el depósito de GL-3 en los ganglios autónomos del intestino y los vasos sanguíneos mesentéricos.5
A pesar de ser frecuentes, los síntomas gastrointestinales siguen siendo una manifestación poco reconocida de EF. Hasta dos tercios de los hombres y la mitad de las mujeres sintomáticas pueden experimentar síntomas gastrointestinales asociados con la enfermedad.5,52,53 Los síntomas gastrointestinales con frecuencia aparecen en la infancia y suelen permanecer presentes durante la edad adulta.
Los pacientes manifiestan dolor abdominal postprandial, diarrea, náuseas y vómitos, que son una causa importante de hiporexia. El síndrome del intestino irritable (SII) con predominio de diarrea es un diagnóstico diferencial.5
La fatiga y el dolor son síntomas de la EF son frecuentes en las afecciones reumatológicas, tales como lupus, fibromialgía y artritis reumatoidea o AIJ 1,2,5,54:
• Dolor articular
• Dolor agudo y crónico de manos o pies, o episodios agudos agonizantes de dolor irradiado en las extremidades que duran de minutos a días ("crisis Fabry")
• Angioqueratomas: lesiones cutáneas de color púrpura rojizo oscuro y pueden parecerse a la vasculitis
• Fiebre recurrente que acompaña al dolor con elevación de ERS
¿Por qué es importante el seguimiento de los pacientes Fabry?



¿No encuentra lo que busca?
Explore nuestro Mapa de sitio.
- Desnick RJ, et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2014:1-64.
- Desnick RJ. Ann Intern Med. 2003;138(4):338-346.
- Laney DA, et al. J Genet Couns. 2008;17(1):79-83
- Eng CM, et al. J Inherit Metab Dis. 2007;30(2):184-192.
- Germain DP. Orphanet J Rare Dis. 2010;5(30):1-49.
- Barba-Romero MA, et al. Int J Clin Pract. 2011;65(8):903-910.
- Mehta A. QJM. 2002;95(10):647-653.
- Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112-128.
- Nagueh SF. Circulation. 2014;130(13):1081-1090.
- Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-1607.
- Sims K. et al. Stroke. 2009:40(3):788-794.
- The Human Gene Mutation Database. Institute of Medical Genetics in Cardiff. Available at: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed January 19, 2018.
- Wang RY et al. Genet Med 2007;9(1):34-45.
- Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-427.
- Germain D. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
- Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, Elliott PM, Linthorst GE, Wijburg FA, Biegstraaten M, Hollak CE. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol. 2017 May;28(5):1631-1641.
- Nakao S, et al. Kidney Int. 2003;64(3):801.807.
- Linthorst GE, et al. Nephrol Dial Transplant. 2003;18(8):1581-1584.
- Kotanako P, et al. J Am Soc Nephrol. 2004;15(5):1323-1329.
- Gupta S, et al. Medicine (Baltimore). 2005;84(5):261-268.
- Shelley ED, et al. Pediatr Dermatol 1995;12(3):215-219.
- Fabry Registry Annual Report 2010. Available at: www.fabry.org/fsig.nsf/PDFs/PDFsR/$File/2010_Annual_Report.pdf. Accessed January 22. 2018.
- Ramaswami U, et al. Clin J Am Soc Nephrol. 2010;5(2):365-370.
- Ichinose M, et al. Clin Exp Nephrol. 2005;9(3):228-232
- Bekri S, et al. Nephrol Clin Pract 2005:101(1):c33-38.
- Coresh J, et al. JAMA. 2007;298(3):2038-2047.
- Waldek S, et al. BMC Nephrol. 2014:15(72):1-15.
- Najafian B, et al. Kidney Int. 2011;79(6):663.670.
- Torra R. Kidney Int. Suppl. 2008;(Suppl. 111):S29-S32.
- Schiffmann R, et al. Nephrol Dial Transplant. 2009.24(7) 2102-2111.
- Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505.517.
- Linthorst GE, et al. J Med Genet. 2010;47(4):217-222.
- Monserrat L, et al. Coll Cardiol. 2007;50(25):2399-2403.
- van der Tol L, et al. J Med Genet. 2014;51(1):1-9.
- Yousef Z, et al. Eur Heart J. 2013;34(11):802-808.
- Eng CM, et al. Genet Med. 2006;8(9):539-548.
- Linhart A, et al. Eur Heart J. 2007;28(10):1228-1235.
- Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-1099.
- Kampmann C, et al. Int J Cardiol. 2008:130(3):367-373.
- Weidemann F, et al. Orphanet J Rare Dis. 2013:8(116).
- Elliott PM, et al. Eur Heart J. 2014;35(39):2733-2779.
- Fellgiebel A, et al. Lancet Neurol. 2006;5(9):791-795.
- Hilz MJ. Clin Ther. 2010;32 (suppl C):S93.
- Bottcher T. et al. Plos ONE. 2013:8(8):e71894.
- Cable WJL, et al. Neurology. 1982;32(5):498-502.
- Uceyler N, et al. Clin J Pain. 2014;30(10):915-920.
- Fellgiebel A, et al. Neurology. 2009;72(1):63-68.
- Moore DF, et al. Brain Res Bull. 2003;62(3):231-240.
- Cole AL, et al. J Inherit Metab Dis. 2007:30(6):943-951.
- Samiy N, et al. Surv Ophthalmol. 2008;53(4): 416-423.
- Hopkin RJ, et al. Pediatr Res. 2008;64(5):550-555.
- MacDermot KD, et al. J Med Genet. 2001a;38(11):750-760.
- MacDermot KD, et al. J Med Genet. 2001b;38(11):769-775.
- Manger B, et al. Clin Rheumatol. 2007;26(3):335-341
Las señales de alerta incluyen dolor neuropático en extremidades (acroparestesias) desde la infancia o adolescencia, angioqueratomas cutáneos, hipohidrosis, intolerancia al calor, opacidades corneales (córnea verticilada), proteinuria o deterioro de la función renal de causa no determinada, hipertrofia ventricular izquierda sin etiología cardiovascular clara, accidentes cerebrovasculares en adultos jóvenes, y antecedentes familiares de enfermedad renal, cardíaca o neurológica. Según las recomendaciones argentinas 2025, el 1% de los pacientes con hipertrofia ventricular izquierda de causa desconocida presenta enfermedad de Fabry.
El diagnóstico se confirma mediante la medición de actividad de alfa-galactosidasa A en leucocitos o en gota de sangre seca (DBS), complementada con el estudio molecular del gen GLA para identificar la variante patogénica. En mujeres, dado que pueden tener actividad enzimática normal por el patrón de inactivación del cromosoma X, el estudio genético es imprescindible para el diagnóstico. El abordaje diagnóstico requiere la colaboración entre nefrólogos, cardiólogos, neurólogos, genetistas clínicos, pediatras, dermatólogos y oftalmólogos.
La nefropatía Fabry es una de las complicaciones más frecuentes y de mayor morbimortalidad. Se manifiesta inicialmente como proteinuria o microalbuminuria, progresando hacia insuficiencia renal crónica que puede requerir diálisis o trasplante alrededor de los 40 años en hombres. La vigilancia recomendada incluye medición de albuminuria y creatinina sérica desde el diagnóstico y con periodicidad anual, evaluación de la tasa de filtrado glomerular y monitoreo de biomarcadores como Gb3 urinario y lisoGb3 plasmático para evaluar la carga de acumulación.
Debes ser un profesional de la salud para continuar leyendo este contenido.
¿Eres un profesional de la salud?
Regístrate para obtener acceso exclusivo a las últimas noticias sobre avances científicos y a recursos que mejoren la vida de tus pacientes.
Login
Obtenga acceso a información para profesionales de la salud y edite los temas relevantes para usted en su perfil.
HCP Verification Required
REENVÍO ÉXITO
Por favor revise su correo electrónico para ver el correo electrónico de verificación.
Contenido restringido
No tienes el rol verificado correcto para ver este contenido.
Revisa tu bandeja de entrada de correo electrónico para activar tu cuenta
Se ha enviado un correo electrónico de verificación al correo electrónico con el que se registró. Para activar su cuenta, haga clic en el enlace de este correo electrónico. Si no ha recibido el correo electrónico de verificación, haga clic en el botón a continuación.
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.