-(1).png/jcr:content.png)
Detrás de cada paciente diagnosticado con Fabry hay una familia que está en riesgo
Patrón hereditario de la enfermedad de Fabry
En la enfermedad de Fabry, las madres afectadas tienen un riesgo del 50% de transmitir el gen GLA mutado a sus hijos independientemente del género, mientras que los padres afectados transmiten el gen GLA defectuoso a todas sus hijas y a ninguno de sus hijos.1
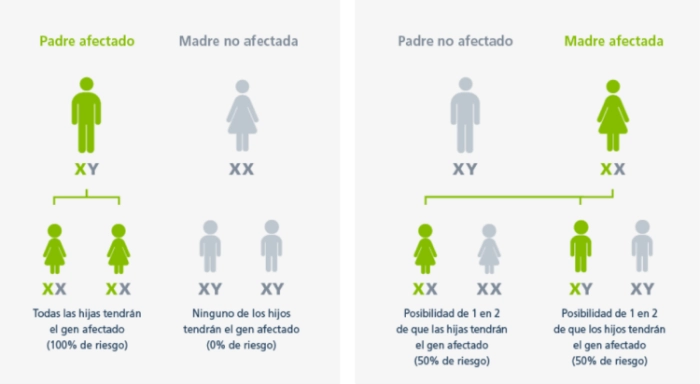
En la enfermedad de Fabry, las madres afectadas tienen un riesgo del 50% de transmitir el gen GLA mutado a sus hijos independientemente del género, mientras que los padres afectados transmiten el gen GLA defectuoso a todas sus hijas y a ninguno de sus hijos.1
La importancia del árbol Genealógico
Una vez que se diagnostica un caso índice de la enfermedad de Fabry, esto posibilita que los familiares pre-sintomáticos puedan ser diagnosticados a una edad más joven, lo que permite un manejo más temprano de la enfermedad. La detección familiar y el análisis de los antecedentes familiares pueden ser especialmente útiles para identificar a las mujeres en riesgo de contraer la enfermedad de Fabry.
La evaluación familiar puede ayudar a encontrar los pacientes en una etapa más temprana en el curso de su enfermedad.

El diagnóstico de cualquier paciente con Fabry debe ser seguido por un estudio genealógico completo en forma de análisis del árbol familiar y la realización de exámenes a los otros miembros de la familia que están en riesgo de la enfermedad de Fabry. Un árbol familiar es el primer paso para identificar a otros miembros de la familia con Fabry.
¿Necesitas asesoramiento?
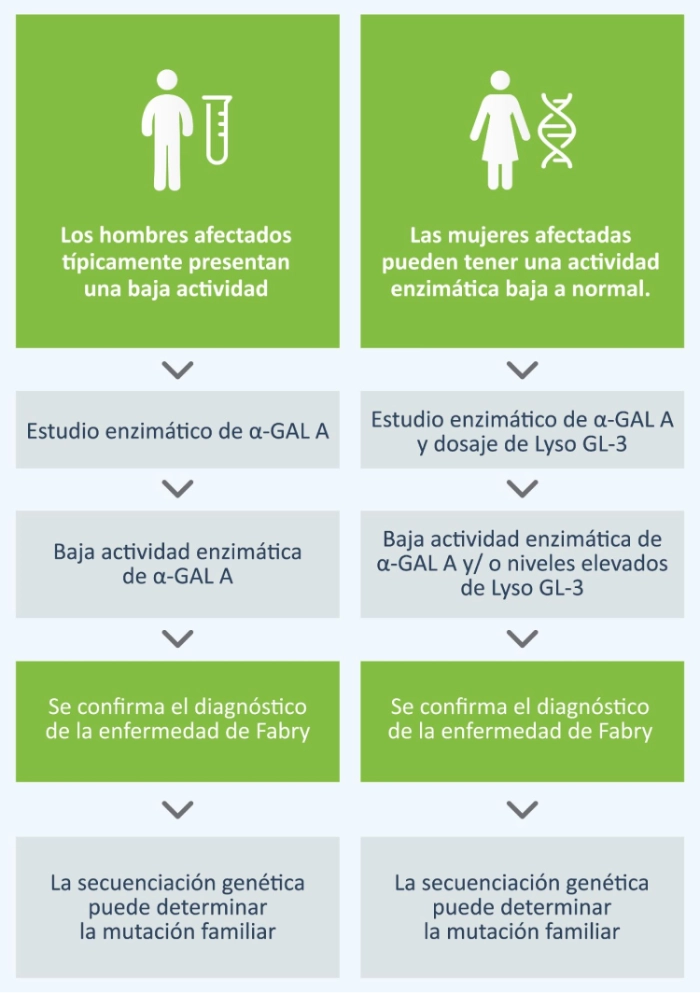
Técnicas diagnósticas
En los hombres, el diagnóstico se realiza mediante la prueba de actividad enzimática de α- galactosidasa A (α- GAL) en plasma o sangre seca. Debido a que las mujeres con enfermedad de Fabry pueden tener actividad en rango bajo a normal se recomienda también realizar el dosaje del biomarcador específico Lyso Gl-3 en gota de sangre seca.
La identificación de una mutación patogénica mediante el estudio del gen GLA es relevante para confirmar el diagnóstico en mujeres y hombres.1 Los resultados pueden ser útiles para comprender las correlaciones entre el genotipo y el fenotipo.
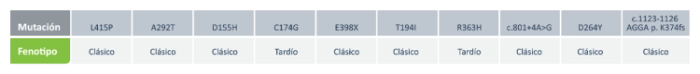
Mutaciones más frecuentes en Argentina
La enfermedad de Fabry se produce a partir de la presencia de una variante patogénica en el gen GLA. Se han identificado más de 900 mutaciones hasta el momento.
Las distintas variantes o mutaciones que existen producen distintos fenotipos: en general los pacientes con enfermedad de Fabry con una actividad enzimática de α-GAL A nula o casi nula (<1%) muestran síntomas severos (fenotipo clásico), mientras que los pacientes con actividad enzimática residual muestran síntomas de aparición más tardía con afectación cardiológica o renal (fenotipo tardío).
Se ha descripto mucha variabilidad fenotípica incluso dentro de una misma familia, sugiriendo que además del genotipo otros factores podrían estar involucrados en la clínica de la enfermedad. Además de los cambios genotípicos (variantes en el gen GLA), se deben considerar los cambios epigenéticos para explicar las diversidades fenotípicas entre los pacientes con las mismas mutaciones.4
Por ende, la enfermedad de Fabry no solo es heterogenética (variabilidad de mutaciones) sino también heterofenotípica. 5 Las 10 mutaciones más prevalentes hoy por hoy en Argentina son las siguientes. Algunas de ellas solo afectan a una sola familia y otras a varias familias en el país y la mayoría producen un fenotipo clásico.
.2023-09-12-11-03-28.webp)
- Barriales-Villa, Gimeno-Blanes, Zorio-Grima, Ripoll-Vera, Evangelista-Masip, Moya-Mitjans, Serratosa-Fernández, Albert-Brotons, García-Pinilla, García-Pavía. Plan of Action for Inherited Cardiovascular Diseases: Synthesis of Recommendations and Action Algorithms. Rev Esp Cardiol 2016;69(3):300-9. doi: 10.1016/j.rec.2015.11.029.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006 Apr 11;113(14):1807-16. DOI: 10.1161/CIRCULATIONAHA.106.174287.
- Perry Elliott, Bert Andersson, Eloisa Arbustini, Zofia Bilinska, Franco Cecchi, Philippe Charron, Olivier Dubourg, Uwe Kühl, Bernhard Maisch, William J. McKenna, Lorenzo Monserrat, Sabine Pankuweit, Claudio Rapezzi, Petar Seferovic, Luigi Tavazzi, Andre Keren. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008;29:270-6. doi.org/10.1093/eurheartj/ehm342 .
- Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 2013 Dec 3;62(22):2046-72. doi: 10.1016/j.jacc.2013.08.1644. Epub 2013 Nov 18. No abstract available. Erratum in: J Am Coll Cardiol 2014 Jan 21;63(2):191-4.
- Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L, Tendera M, Maupain C, et al. EORP Cardiomyopathy Registry Investigators. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J 2018;39(20):1784-1793. doi: 10.1093/eurheartj/ehx819.
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/America
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.