- Article
- Source: Campus Sanofi
Testeo y diagnóstico de la enfermedad de Pompe: pruebas enzimáticas y genéticas
Guía clínica sobre el proceso de testeo y diagnóstico de la enfermedad de Pompe, incluyendo la medición de actividad de alfa-glucosidasa ácida (GAA) en gota de sangre seca (DBS), confirmación en leucocitos o fibroblastos, estudio molecular del gen GAA, y criterios de interpretación para las formas infantil y de inicio tardío.
El primer paso es la medición de actividad de GAA mediante gota de sangre seca en papel de filtro (DBS), un método de alta sensibilidad desarrollado en Argentina. Un resultado de actividad reducida debe confirmarse con medición enzimática en leucocitos circulantes (método estándar) o en fibroblastos cultivados (más invasivo, reservado para casos dudosos). La confirmación definitiva se realiza mediante estudio molecular del gen GAA, que identifica las variantes patogénicas bialélicas. La base de datos del Erasmus Center (Pompe Variant Database) con 911 variantes es la referencia internacional para interpretar los resultados.
Un resultado de actividad de GAA reducida en DBS tiene alta sensibilidad para la enfermedad de Pompe pero requiere confirmación, ya que pueden existir falsos positivos por pseudodeficiencia (variantes benignas del gen GAA que reducen la actividad enzimática sin causar enfermedad). La confirmación enzimática en leucocitos establece si la actividad es significativamente deficiente (generalmente <1% en forma infantil y entre 1–40% en inicio tardío). El estudio genético del gen GAA permite distinguir variantes patogénicas de pseudodeficiencia y orientar el consejo genético familiar.
El DBS tiene sensibilidad cercana al 100% para la forma infantil clásica y alta sensibilidad para la forma tardía en la mayoría de los casos. Su especificidad es moderada por la presencia de pseudodeficiencia, por lo que los resultados positivos siempre requieren confirmación. El tamizaje por DBS está indicado en adultos con debilidad muscular proximal de causa no determinada, compromiso respiratorio de origen muscular, o en el contexto de programas de cribado neonatal para detección precoz. Su bajo costo, no invasividad y facilidad de recolección lo hacen adecuado para el primer nivel diagnóstico.
Diagnóstico diferencial
Distinguir la enfermedad de Pompe de otros trastornos es fundamental para minimizar los retrasos en el diagnóstico y optimizar los resultados de los pacientes
Los pacientes con LOPD pueden experimentar una demora de entre 6 a 13 años antes de recibir un diagnóstico definitivo, y los pacientes con enfermedad de Pompe de inicio infantil (IOPD) experimentan una demora media de 1,4 meses desde el momento de inicio de los síntomas hasta el diagnóstico. El deterioro clínico durante estas brechas diagnósticas puede ser significativo debido al avance del daño irreversible y potencialmente mortal.
El diagnóstico y el tratamiento precoces son clave para controlar los síntomas y mejorar los resultados en pacientes con enfermedad de Pompe. Sospeche la enfermedad de Pompe en bebés, niños y adultos cuando los signos y síntomas sugieran una degeneración muscular progresiva.
Sospeche la enfermedad de Pompe cuando observe alguno de los siguientes signos o síntomas:
.2023-02-27-11-36-55.2024-04-25-15-49-17.png)
- Debilidad en las piernas
- Dificultad para subir escaleras
- Dificultad para levantarse de una silla
- Caminar balanceando las caderas o rengueando
- Caídas frecuentes y problemas para correr o hacer deporte
.2023-02-27-11-37-53.2024-04-25-15-50-51.png)
- Disnea al hacer esfuerzo, p. ej. subir escaleras
- Dificultad para respirar, especialmente en decúbito dorsal
- Enfermedad pulmonar restrictiva y tos inefectiva
- Dificultad respiratoria nocturna, manifestada por apnea del sueño, dolores de cabeza matutinos o niveles elevados de CO2
- Hipoventilación durante el sueño
- Fatiga excesiva durante el día
- Intolerancia al ejercicio
Otras enfermedades y trastornos pueden manifestarse con signos y síntomas similares a la enfermedad de Pompe; incluya la enfermedad de Pompe en su diagnóstico diferencial junto con aquellas con cuadros clínicos similares

Superposición de síntomas entre LOPD y otros trastornos más comunes

Superposición de síntomas entre IOPD y otros trastornos más comunes
Una vez que surge la sospecha clínica, se puede evaluar la enfermedad de Pompe midiendo la actividad de la enzima α-glucosidasa ácida (GAA)
Considere la posibilidad de realizar pruebas para detectar la enfermedad de Pompe cuando vea
- Debilidad progresiva de los músculos proximales con o sin insuficiencia respiratoria o hiperCKemia persistente inexplicable (niveles leves a moderadamente elevados, ~300-2000 U/L)
- o Los niveles normales de CK no descartan la enfermedad de Pompe
- Miopatía de cintura y extremidades no especifica
- Un miembro de la familia que padece la enfermedad de Pompe
¿Podría la enfermedad de Pompe pasar inadvertida en su consultorio?
Obtenga más información sobre los signos y síntomas que afectan a los pacientes que padecen la enfermedad de Pompe.
Función de las imágenes por resonancia magnética (RM) en el diagnóstico
Los resultados de estudios en pacientes con LOPD han demostrado una correlación entre la debilidad muscular y los hallazgos anormales de las RM y la tomografía computarizada
Las técnicas de imagen avanzadas como la RM pueden identificar la atrofia y la infiltración grasa de músculos específicos, así como patrones de afectación muscular, aunque no son exclusivas de la enfermedad de Pompe y no establecen un diagnóstico definitivo.
En un estudio de historia natural, los pacientes con LOPD tenían deficiencias significativas en la expansión de los pulmones durante la inspiración en comparación con los voluntarios sanos (P = 0,001)
Las reducciones en el volumen inspiratorio máximo tienen un impacto directo y negativo en la función respiratoria al limitar la capacidad vital forzada (CVF)
En el mismo estudio, los pacientes con LOPD mostraron una disminución anual significativa de la función respiratoria:
- Disminución anual del 3,2 % en la presión inspiratoria máxima (P = 0,018)
- Disminución anual del 3,8 % en la presión espiratoria máxima (P <0,01)
Pulmones sanos Enfermedad de Pompe
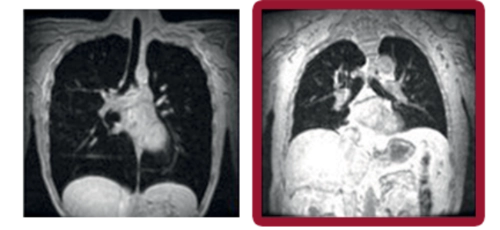
Volumen inspiratorio máximo
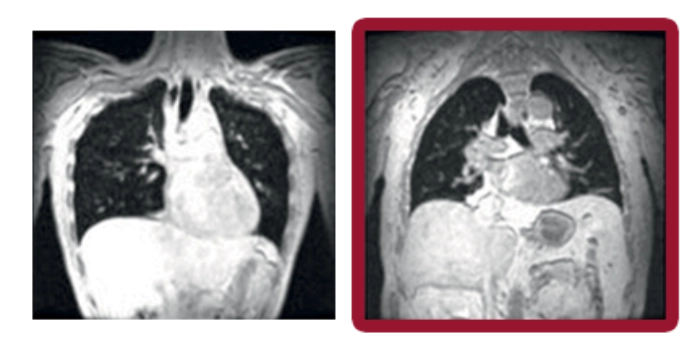
Volumen inspiratorio máximo
Las imágenes coronales de resonancia magnética evaluaron los movimientos máximos de final de inspiración y final de espiración mientras los sujetos contenían la respiración durante 12 segundos. El volumen pulmonar (representado en negro) en cada sujeto se calculó fuera de línea y se normalizó por la proporción de la longitud del pulmón en la inspiración dividida por la longitud en la espiración.
¿Podría la enfermedad de Pompe pasar inadvertida en su consultorio?
Obtenga más información sobre los signos y síntomas que afectan a los pacientes que padecen la enfermedad de Pompe.
Función de la electromiografía (EMG) en el diagnóstico
Los estudios demuestran que más del 70 % de los pacientes con LOPD tienen un patrón de EMG miopático
Resultado de la EMG que sugiere LOPD
- Irritabilidad notablemente aumentada de la membrana muscular
- o Potenciales de fibrilación
- o Ondas agudas positivas
- o Descargas repetitivas complejas
- Descargas miotónicas (típicas o atípicas)
- Acción breve de la unidad motora (potenciales de acción de la unidad motora)
Los resultados de la EMG en los músculos más proximales, incluidos los músculos paraespinales, son más propensos a mostrar anomalías.
La EMG debe realizarse en los músculos proximales y distales de las extremidades superiores e inferiores, así como en los músculos paraespinales torácicos si se sospecha LOPD
Diagnóstico definitivo
Secuencia diagnóstica
Aunque las secuencias clínicas para el diagnóstico de la enfermedad de Pompe son variables, el proceso generalmente implica:

Evaluación clínica de los síntomas
presentes por un médico clínico

Derivación a un especialista para una
investigación más profunda, incluidas
pruebas clínicas o de laboratorio adicionales

Evaluación clínica de los síntomas
presentes por un médico clínico
.2024-04-25-17-40-16.svg)
Las pruebas de actividad de la enzima GAA pueden identificar a los pacientes que tienen la enfermedad de Pompe
Prueba de gota de sangre seca (DBS, por sus siglas en inglés)
La enfermedad de Pompe se confirma por una ausencia completa o una reducción marcada de la actividad de GAA.
El uso de muestras de sangre, incluida la DBS, es ahora una práctica estándar. La toma de muestras de sangre para las pruebas de detección de la enfermedad de Pompe es mínimamente invasiva, precisa y, por lo general, puede proporcionar resultados en unos pocos días. Si se encuentra una actividad reducida de la enzima GAA, el diagnóstico debe confirmarse en una segunda muestra (ej leucocitos) o mediante un análisis de mutación para confirmar la presencia de 2 alelos del gen GAA mutado.
Históricamente, el informe de la enzima GAA se realizó usando fibroblastos cutáneos cultivados. Sin embargo, la recolección de muestras es relativamente invasiva y los resultados tardan aproximadamente 6 semanas en obtenerse. Este largo tiempo de respuesta no es recomendable, especialmente en bebés con enfermedad rápidamente progresiva.
Aunque las biopsias musculares son una opción para las pruebas de actividad de GAA, generalmente no son las preferidas. Esto se debe a que son invasivas y tienen un alto riesgo de falsos positivos debido al mal manejo de las muestras. Las biopsias musculares pueden ser útiles para la evaluación histológica, pero es importante tener en cuenta que el contenido de glucógeno puede variar ampliamente entre los músculos, por lo que las biopsias aparentemente normales no descartan la enfermedad de Pompe. Por lo tanto, un diagnóstico de enfermedad de Pompe siempre debe confirmarse mediante la ausencia o reducción de la actividad de GAA o mediante análisis genético.

En pacientes con IOPD, la actividad de la enzima GAA puede ser completamente nula; si queda algo de actividad, suele ser <1 % de lo normal

En pacientes con LOPD, la actividad de la enzima GAA suele estar entre el 1 % y el 40 % de lo normal
Para los pacientes diagnosticados con la enfermedad de Pompe, el control precoz y activo es esencial
La atención interdisciplinaria continua puede ayudar a mejorar los resultados en pacientes con enfermedad de Pompe.
Pruebas de detección a familiares
La enfermedad de Pompe es una afección hereditaria que puede transmitirse de padres a hijos
Si bien la enfermedad de Pompe se considera rara en la población general, los parientes cercanos de una persona con la enfermedad de Pompe tienen muchas más probabilidades de padecer la enfermedad o ser portadores. Las pruebas de detección a familiares de pacientes con enfermedad de Pompe puede ayudar en el diagnóstico temprano y en un control más eficaz de la enfermedad.
References: 1. Kishnani PS, et al; on behalf of the Pompe Registry Boards of Advisors. Am J Med Genet. 2013;161A(10):2431-2443. 2. Kishnani PS, et al; ACMG Work Group on Management of Pompe Disease. Genet Med. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3. 3. Kishnani PS, et al. J Pediatr. 2004;144(5 suppl):S35-S43. 4. Rigter T, et al. Mol Genet Metab. 2012;107(3):448-455. doi:0.1016/j.ymgme.2012.09.017. 5. Schüller A, et al. Am J Med Genet C Semin Med Genet. 2012;160C(1):80-88. doi:10.1002/ajmg.c.31322. 6. van Capelle CI, et al. Orphanet J Rare Dis. 2016;11(1):65. doi:10.1186/s13023-016-0442-y. 7. American Association of Neuromuscular and Electrodiagnostic Medicine. Muscle Nerve. 2009;40(1):149-160. doi:10.1002/mus.21393. 8. Kishnani PS, et al; Infantile-Onset Pompe Disease Natural History Study Group. J Pediatr. 2006;148(5):671-676. doi:10.1016/j.jpeds.2005.11.033. 9. Hirschhorn R, et al. In: Scriver CR, et al, eds. The Metabolic Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420. 10. Fusco AF, et al. Int J Mol Sci. 2020;21(6):2256. doi:10.3390/ijms21062256. 11. Hereditary myopathy with early respiratory failure. Genetics Home Reference website. Hereditary myopathy with early respiratory failure: MedlinePlus Genetics . Reviewed September 2018. Accessed December 15, 2020. 12. Barohn RJ, et al. Neurol Clin. 2014;32(3):569-vii. doi:10.1016/j.ncl.2014.04.008. 13. Chaudhuri A, et al. Lancet. 2004;363(9413):978-988. doi:10.1016/S0140- 6736(04)15794-2. 14. Jaradeh S. Muscle disorders affecting oral and pharyngeal swallowing. GI Motility Online website. Muscle disorders affecting oral and pharyngeal swallowing . Published May 16, 2006. Accessed December 15, 2020. 15. Gilchrist JM. Semin Respir Crit Care Med. 2002;23(3):191-200. doi:10.1055/s-2002-33027. 16. Ozawa E, et al. Mol Cell Biochem. 1999;190:143-151. 17. Mah JK, et al. Neuromuscul Disord. 2014;24(6):482-491. doi:10.1016/j.nmd.2014.03.008. 18. Barnabei MS, et al. Compr Physiol. 2011;1(3):1353-1363. doi:10.1002/cphy.c100062. 19. Limb-girdle muscular dystrophy. Genetics Home Reference website. Limb-girdle muscular dystrophy: MedlinePlus Genetics . Reviewed September 2019. Accessed December 15, 2020. 20. Pegoraro E, et al. In: Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. NCBI - WWW Error Blocked Diagnostic . Published June 8, 2000. Updated August 30, 2012. Accessed December 15, 2020. 21. Siciliano G, et al. Acta Myol. 2015;34(1):3-8. 22. Myasthenia gravis. Genetics Home Reference website. https://ghr.nlm.nih.gov/condition/myastheniagravis. Published February 27, 2017. Accessed December 15, 2020. 23. Myasthenia gravis: what is it? Harvard Health Publishing website. https://www.health.harvard.edu/a_to_z/myasthenia- gravis-a-to-z. Published December 2018. Accessed December 15, 2020. 24. Smoyer-Tomic KE, et al. BMC Musculoskeletal Disord. 2012;13:103. doi:10.1186/1471- 2474-13-103. 25. Inflammatory myopathies information page. National Institute of Neurological Disorders and Stroke website. Inflammatory Myopathies . Published February 27, 2017. Accessed December 15, 2020. 26. Gazeley DJ, et al. Ther Adv Musculoskeletal Disord. 2011;3(6):315-324. doi:10.1177/1759720X11415306. 27. Polymyositis. Johns Hopkins Medicine website. Polymyositis . Published February 27, 2017. Accessed December 15, 2020. 28. Mastaglia FL, et al. Rheum Dis Clin North Am. 2002;28(4):723-741. doi:10.1016/s0889-857x(02)00021-2. 29. Rudnik-Schöneborn S, et al. Eur Neurol. 1998;39(3):154-162. doi:10.1159/000007926. 30.Davis RH, et al. J Child Neurol. 2014;29(11):1467-1472. doi:10.1177/0883073813503988. 31.D'Souza RS, et al. Circ Heart Fail. 2014;7(5):843-849. doi:10.1161/CIRCHEARTFAILURE.114.001105. 32. Fu L, et al. Korean Circ J. 2013;43(12):785-792. doi:10.4070/kcj.2013.43.12.785. 33. Leslie N, et al. GeneReviews. NCBI - WWW Error Blocked Diagnostic . Published August 31, 2007. Updated May 11, 2017. Accessed December 15, 2020. 34. Toscano A, et al. Acta Myol. 2013;32(2):78-81. 35. Preisler N, et al. Mol Genet Metab. 2013;110(3):287-289. doi:10.1016/j.ymgme.2013.08.005. 36. Manganelli F, et al. Acta Myol. 2013;32(2):82-84. 37. Moghadam-Kia S, et al. Cleve Clin J Med. 2016;83(1):37-42. doi:10.3949/ccjm.83a.14120. 38. Wens SCA, et al. BMC Pulmonary Medicine. 2015;15:54. doi:10.1186/s12890-015-0058-3. 39. Figueroa-Bonaparte S, et al. PLoS One. 2016;11(10):e0163493. doi:10.1371/journal.pone.0163493. 40. Paoletti M, et al. Front Neurol. 2019;10:78. doi:10.3389/fneur.2019.00078. 41. van der Beek NAME, et al. Mol Genet Metab. 2011;104:129-136. 42. Müller-Felber W, et al. Neuromuscul Disord. 2007;17(9-10):698-706. doi:10.1016/j.nmd.2007.06.002. 43. Wagner M, et al. Neuromuscul Disord. 2013;23(1):89-92. doi:10.1016/j.nmd.2012.09.004. 44. Behjati S, et al. Arch Dis Child Educ Pract Ed. 2013;98(6):236-238. doi:10.1136/archdischild-2013-304340. 45. Okumiya T, et al. Mol Genet Metab. 2006;88(1):22-28. doi:10.1016/j.ymgme.2005.10.016. 46. Cupler EJ, et al; AANEM Consensus Committee on Late-Onset Pompe Disease. Muscle Nerve. 2012;45(3):319-333. doi:10.1002/mus.22329. 47. Bodamer OA, et al; on behalf of the Pompe Disease Newborn Screening Working Group. Pediatrics. 2017;140(suppl 1):S4-S13. doi:10.1542/peds.2016-0280C. 48. Recommended uniform screening panel. Health Resources and Services Administration website. Recommended Uniform Screening Panel | HRSA . Reviewed February 2020. Accessed December 15, 2020. 49. Taverna S, et al. Aging (Albany NY). 2020;12(15):15856-15874. doi:10.18632/aging.103794. 50. Atherton AM, et al. Pediatrics. 2017;140(suppl 1):S46-S50. doi: 10.1542/peds.2016-0280F. 51. Winchester B, et al. Mol Genet Metab. 2008;93(3):275-281. doi: 10.1016/j.ymgme.2007.09.006. 52. Zhang H, et al. Genet Med. 2006;8(5):302-306. doi: 10.1097/01.gim.0000217781.66786.9b. 53. van der Ploeg AT, et al. Eur J Neurol. 2017;24(6):768-e31. doi:10.1111/ene.13285. 54. Llerena JC Jr, et al. Arq Neuropsiquiatr. 2016;74(2):166-176. doi:10.1590/0004-282X20150194.
Áreas de evaluación
Áreas clave para el seguimiento de la enfermedad de Pompe
- Sin tratamiento, la mayoría muere por insuficiencia cardiorrespiratoria al año de edad
- Radiografías de tórax y ecocardiograma (ECG) para evaluar la miocardiopatía
- Holter de 24 horas para detección de arritmias
- Monitoreo del peso y del crecimiento
- Evaluación física
- Evaluaciones de calidad de vida (bienestar mental y emocional)
- La insuficiencia respiratoria es la causa más común de muerte entre los pacientes con enfermedad de Pompe
- Pruebas de rutina de la función pulmonar (p. ej., capacidad vital pulmonar y fuerza diafragmática)
- Historial de sueño detallado y seguimiento continuo de trastornos respiratorios del sueño
- Evaluaciones según criterio médico ej radiografía, electromiografía y puntos de referencia de hitos motores.
- Densitometría. Evaluación metabolismo óseo
Debes ser un profesional de la salud para continuar leyendo este contenido.
¿Eres un profesional de la salud?
Regístrate para obtener acceso exclusivo a las últimas noticias sobre avances científicos y a recursos que mejoren la vida de tus pacientes.
Login
Obtenga acceso a información para profesionales de la salud y edite los temas relevantes para usted en su perfil.
HCP Verification Required
REENVÍO ÉXITO
Por favor revise su correo electrónico para ver el correo electrónico de verificación.
Contenido restringido
No tienes el rol verificado correcto para ver este contenido.
Revisa tu bandeja de entrada de correo electrónico para activar tu cuenta
Se ha enviado un correo electrónico de verificación al correo electrónico con el que se registró. Para activar su cuenta, haga clic en el enlace de este correo electrónico. Si no ha recibido el correo electrónico de verificación, haga clic en el botón a continuación.
.png/jcr:content/BALANCED%20(2).png)
ICNMD 2022 Pompe Symposium - Jordi Díaz Manera Slides
External advisoy board of Sanofi- Genzyme, Audentes and Lupin, Received honoraria for lectures and workshops fron Sanofi- Genzyme
.png/jcr:content/MAIN-TEXT%20(2).png)
PMP-CAT3-STP4-01 Registro español de la enfermedad de Pompe - Dr. Rafael Jenaro
Glososis tipo dos, es una enfermedad en la cual se observan acúmulos de Glucógeno. Esta esta enfermedad ha sido se han observado más de 350 acotaciones patogénicas entre las conocidas en Exones e Intrones

Que la suspensión de la terapia sea la ultima opción
Que la suspensión de la terapia sea la última opción. No se debe pensar que la suspensión de la terapia sea la primera opción y ante una adversidad que se nos presente, es posible echar mano de otras opciones, ya que la suspensión es mucho más compleja de lo que se piensa (Estudio Suiza).
perfiles-de-pacientes-lucas
.2024-03-12-15-52-14.svg)
Perfil clínico
- 16 años, diagnosticado hace 8 año
- Sufre de signos y síntomas intensos.
- Sufre de signos y síntomas intensos.
- Tratamiento actual: CET de alta potencia. Sin respuesta a fototerapia.
Impacto
- Las lesiones altamente visibles en el rostro tienen un impacto en su vida social.
- El prurito lo distrae durante las clases y no lo deja dormir por las noches.
- Tuvo siete ausencias escolares este año debido a la enfermedad.
- La enfermedad tiene, además, un impacto negativo en sus padres, incluido el estrés.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Distraerse menos en la escuela a causa de sus síntomas.
perfiles-de-pacientes-sofia

Perfil clínico
- 26 años, diagnosticada en la primera infancia.
- Ha sufrido erupciones problemáticas y trastornos frecuentes del sueño.
- Mejora clínica inadecuada con el tratamiento actual.
- Tratamiento actual: CET. Debió suspender ciclosporina por efectos adversos.
Impacto
- Lesiones cutáneas en las áreas de flexión.
- El prurito persistente provoca trastornos del sueño.
- El prurito persistente provoca trastornos del sueño.
Metas del tratamiento
- Reducir el prurito.
- Mejorar las lesiones cutáneas.
- Encontrar un tratamiento que permita un control adecuado a largo plazo de su enfermedad sin toxicidad.
perfiles-de-pacientes-mateo

Perfil clínico
- 50 años, diagnosticado en la primera infancia.
- No logra sostener el control a largo plazo de la enfermedad con el tratamiento actual
- Tratamiento actual: CET y ciclosporina, con pobre respuesta.
Impacto
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.
Metas del tratamiento
- Lesiones crónicas: liquenificación con sobre-infecciones frecuentes.
- Prurito severo persistente.
- La pérdida frecuente del sueño afecta el desempeño laboral.