Fabry disease
Fabry disease is an X-linked lysosomal storage disease due to a defect in the gene encoding the lysosomal enzyme alpha-galactosidase A (α-Gal A), causing progressive cellular accumulation of the substrate globotriaosylceramide (GL-3) and globo-triaosylsphingosine (lyso-GL-3).

Fabry disease is an X-linked lysosomal storage disease due to a defect in the gene encoding the lysosomal enzyme alpha-galactosidase A (α-Gal A), causing progressive cellular accumulation of the substrate globotriaosylceramide (GL-3) and globo-triaosylsphingosine (lyso-GL-3).
This accumulation occurs in a variety of cell types and can lead to debilitating symptoms such as neurological pain, angiokeratoma, hypohidrosis in childhood, in girls usually a few years later than in boys. With age, progressive damage to the vital organs develops in both sexes that leads to organ failure. End-stage kidney disease and life-threatening cardiovascular or cerebrovascular complications limit life expectancy.
Although the disease is X-linked, most women develop symptoms. Fabry disease is pan-ethnic. Newborn screenings report frequencies of 1 in 22,570 men for the classic phenotype and of 1 in 1,390 men for the late-onset phenotype.1-3

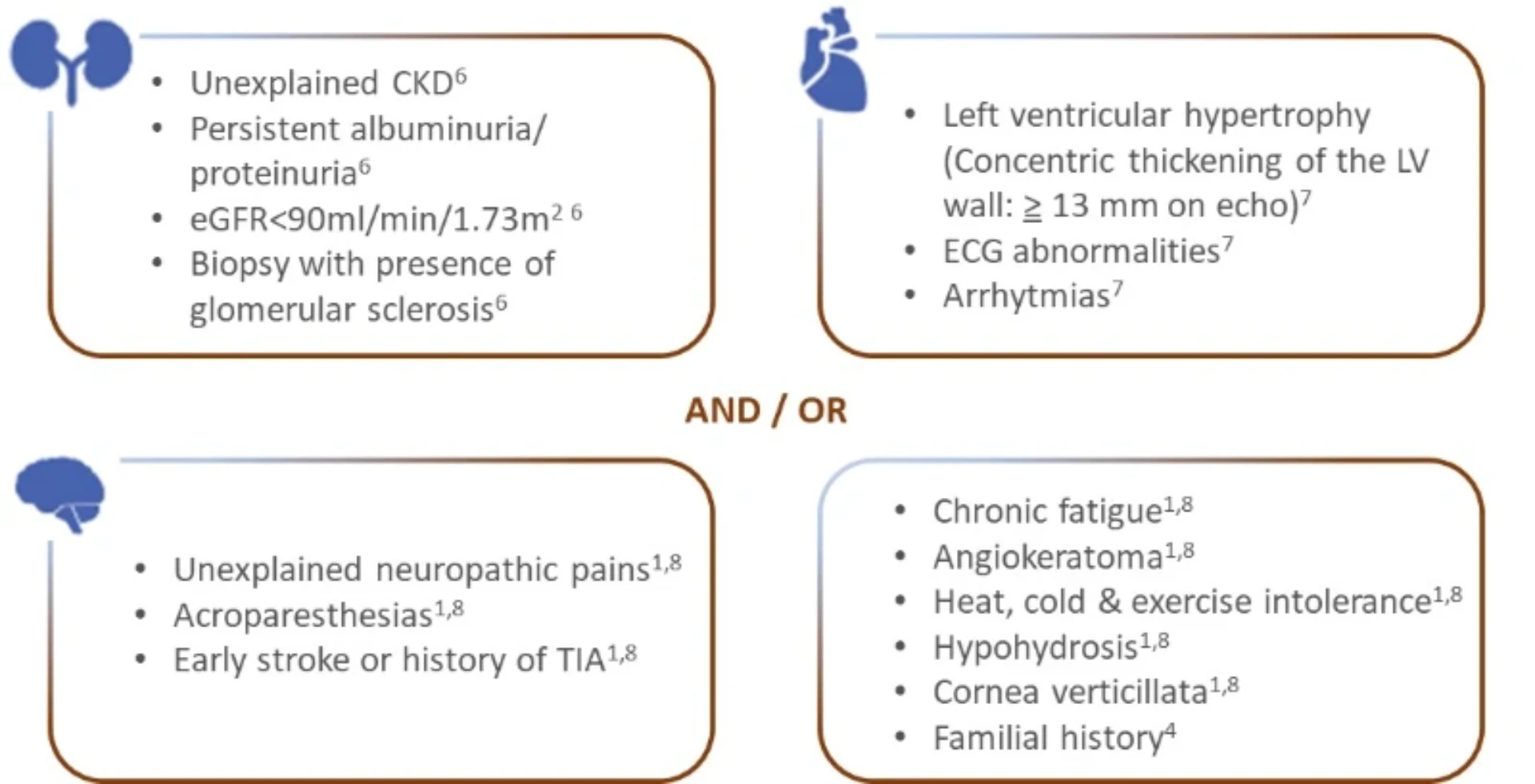
Suspect Fabry disease in case of..
Test the patient, Test the family & Treat early

Up to 5 family members may be diagnosed with Fabry disease for every index patient4
.png)
Lyssna på KardiologKvarten podcast
En podd med fokus på hypertrofisk kardiomyopati (HCM) och riktar sig till dig som möter patienter med HCM. Varje avsnitt är cirka 15 minuter långa.
Lyssna på KardiologKvarten här!
References
-
Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30
-
Germain D., Fabry Disease. Orphanet encyclopedia, March 2022, http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=324
-
Ortiz A, Germain DP, Desnick RJ et al. Fabry disease revisited: management and treatment recommendations for adult patients. Molecular genetics and metabolism 123.4 (2018): 416-427
-
Laney DA & Fernhoff PM. Diagnosis of Fabry Disease via Analysis of Family History. J Genet Couns. 2008; 17(1):79-83.
-
MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001, 38(11):769-775
-
Terryn W. Cochat P, Froissart R et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice Nephrol. Dial. Transplant. 2013, 28: 505-517,
-
Yousef Z, Elliott PM, Cecchi F et al. Left ventricular hypertrophy in Fabry disease: a practical approach to diagnosis. Eur Heart J. 2013, 34:802-808,
-
Eng CM, Germain DP, Banikazemiet M et al. Fabry disease: Guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539-48

How to diagnose Fabry disease
The examination of α-Gal A enzymatic activity can be performed on a Dried Blood Spot (DBS) test. Determination of pronounced α-Gal A deficiency is the definitive method of diagnosis in men.

Building Care webinar series
The Building Care webinar series is designed to deepen your understanding of the critical role a patient-centered, multidisciplinary approach plays in managing Fabry disease.

A Heart for Fabry - Podcast
A podcast series designed for cardiologists and healthcare professionals, focusing on the critical role of cardiologists in diagnosing and managing Fabry disease.
SE-FILMEN-HÄR
